

Businesses often overlook the power of well-executed, competent complaint-handling systems in boosting business growth. Data from ABa Quality Monitoring Ltd revealed nearly 70% of customers are more likely to stick with a company if their complaints are resolved, and this figure jumps to an impressive 95% if complaints are addressed immediately.
Every company faces distinct operational challenges that can result in missed deadlines, product holds, supply chain disruptions and customer complaints. Each of which can impact the quality of the enterprise’s product or service.
This is why the ability to conduct a comprehensive root cause analysis (RCA) to identify the origin of concerns and formulate and execute remedial processes, such as corrective and preventive actions (CAPA), is essential for food companies. Root cause analyses reduce the risk of future nonconformances and complaints and help streamline business operations and protect your bottom line.
An Overview of CAPA and Root Cause Analysis
Corrective and Preventive Action, or CAPA, is a methodical system used across industries to identify, rectify and avert issues, errors or noncompliance in processes, products or systems. Corrective actions fix existing problems, while preventive actions stop their recurrence.
Root Cause Analysis (RCA) encompasses various problem-solving methodologies, permitting teams to identify the fundamental reasons behind issues. It goes beyond just detecting the main problem to determine contributing factors, implement corrective and preventive measures, and foster continuous improvement in business processes.
Conducting a Root Cause Analysis
A CAPA management system’s success hinges on the RCA strategy’s efficiency. Whether businesses are facing a manufacturing defect, a customer complaint, a safety incident or any other problem, conducting an RCA is instrumental in identifying the underlying reasons and implementing effective solutions. Rather than relying on a one-size-fits-all approach, RCA should comprise a highly adaptable toolkit of techniques, and methods. Here are the basic steps for performing a root cause analysis:
Step 1: Understand the Issue
Initiate the process by clearly defining the problem or incident that must be examined. Ensure that the problem statement is specific, measurable and well-documented, enabling a precise understanding of what needs investigation.
Step 2: Collect Information
Aggregate all available data related to the issue, which may include incident reports, witness statements, photographs, process documentation and any other pertinent information. The more data you collect, the better your grasp of the problem.
Step 3: Identify Root Causes
Recognize the immediate or proximate factors that directly contributed to the problem. These are the most visible elements that directly played a role in the incident. Beyond these immediate causes are the contributing factors, often systemic, which may have indirectly influenced the problem. Thoroughly investigate and unearth the foundational reasons behind the issue using appropriate RCA techniques or methods.
Step 4: Develop Corrective and Preventive Actions
Once the root causes are uncovered, brainstorm and assess suitable corrective and preventive actions. Prioritize these solutions based on their practicality, impact and cost-effectiveness, and put the chosen solutions into effect.
Step 5: Establish a Coordinated Plan
Be sure to establish a plan for carrying out the corrective and preventive actions. This plan should include details about the required resources, the assignment of responsible individuals for task execution and an evaluation of the potential risks associated with these corrective measures.
Step 6: Monitor and Verify
Continuously monitor the situation to ensure that the problem does not reoccur. This may involve ongoing training, regular audits or process improvements.
Methods for Root Cause Analysis
Each root cause analysis method offers a unique approach to uncovering the root cause of a problem, providing a systematized way to investigate and address issues. Following are root cause analysis techniques that are adaptable for standalone or combined use, determined by the issue’s intricacy and the particular objectives of the RCA.
- The 5 Whys: This technique involves asking “why” multiple times to dig deeper into a problem. Repeatedly questioning the cause behind a situation helps uncover the underlying issues. The goals is to identify the fundamental reason and avoid assumptions.
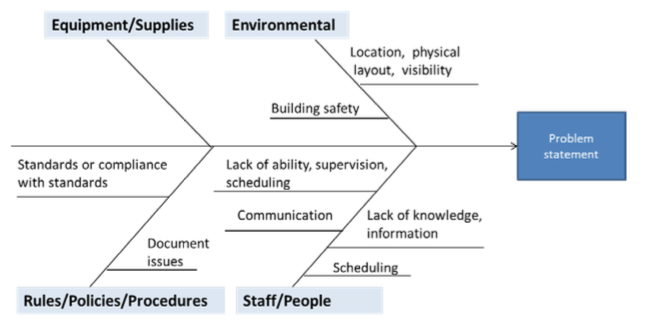
- Fishbone Diagram: This visual tool organizes potential causes of a problem into categories, such as people, processes, equipment, environment and materials. It’s great for exploring multiple factors contributing to an issue.
- FMEA (Failure Modes and Effects Analysis): This method evaluates potential failure modes and their impacts before they occur. It helps in identifying failure points and assessing the existing controls to address these issues.
- DMAIC (Define, Measure, Analyze, Improve, Control): A data-driven improvement cycle, dividing the problem-solving process into five steps: defining the problem, collecting data, analyzing root causes, putting solutions into action and establishing controls for sustained improvement.
- Pareto Analysis: Based on the Pareto Principle, it suggests that a significant portion of issues (80%) can be linked to fewer causes (20%). This tool helps prioritize efforts and resources for problem-solving by focusing on the vital few causes.
- FTA (Fault Tree Analysis): This deductive method works backward from an undesirable event, using graphical representations to analyze relationships between various events and their causes. It uses boolean logic to determine root causes.
Tech-Enabled Solutions to Optimize RCA Processes
In a world where time-consuming manual tasks can hinder productivity, embracing CAPA management and root cause analysis process technology can drive excellence across your enterprise. By digitalizing CAPA processes, organizations can seamlessly manage the entire lifecycle of complaints, holds and deviations.
Digital systems and software for RCA and CAPA management can enable food companies to centralize and automate the recording and tracking of customer complaints, product holds and deviations from established standards, which will evidently lead to faster response times, improved data analysis and better communication across departments. These digital tools aid in identifying recurring issues for swift remediation while preventing similar problems in the future by facilitating real-time monitoring and reporting.
An ideal CAPA management solution will possess the following digital functionalities to revolutionize the process:
Configurable Record Templates. Customized templates for recording complaints, deviations, holds and CAPA can be created or tailored flexibly, allowing users to accommodate differing data requirements to solve a problem. This helps standardize and streamline the documentation processes involved in RCA and CAPA procedures, as these templates help in the consistent recording of incident details, root causes, corrective actions and preventive measures, simplifying the generation of auditable records. Not to mention, they would play a significant role in reducing data entry errors and saving time, consequently supporting a proficient CAPA management system.
CAPA Workflow Automation. CAPA management tools are designed to facilitate the automation of various stages of CAPA, from incident reporting to the review and approval process. By automating tasks such as routing for reviews, approvals and monitoring deadlines for corrective actions, workflow automation significantly reduces the need for manual intervention, saving time and minimizing the potential for human error within the workflow. As a result, the entire CAPA process becomes less resource-intensive and more refined, ensuring a more seamless journey from identification to resolution.
Investigation & Root Cause Analysis. Digitalization ushers a thorough investigation process by collecting documentary evidence in an organized manner, enabling a quicker and more accurate determination of root causes and leading to more efficient corrective and preventive actions to prevent recurrence.
Integrated systematic RCA models into the digital system expedites the root cause analysis process while amplifying its efficacy. Through continuous data gathering from machinery and processes, advanced analytics platforms identify patterns and anomalies, aiding in pinpointing the root cause of failures or issues. Visualization tools provide clear insights into complex relationships among various factors, aiding in understanding causality and devising adequate solutions.
Collaboration is also substantially improved through digital platforms, allowing multidisciplinary teams to collaborate seamlessly regardless of their locations. These tools help structure the RCA process, ensuring tasks are assigned, progress is monitored, and deadlines are met, thus driving the investigation into a more optimal and exhaustive RCA analysis.
Non-Compliance Management. Well-defined planning and implementation of noncompliance action items become possible through digital tools by assigning corrective and preventive actions to respective stakeholders for prompt rectification, compliance maintenance and future risk mitigation. Furthermore, evaluating and validating the effectiveness of corrective and preventive actions is a critical step in RCA. Ensuring the successful closure of the CAPA process by reviewing and confirming the resolution of nonconformances becomes more efficient with digital solutions.
Notification & Alerts. CAPA management systems can be configured to send timely notifications and alerts to relevant stakeholders and responsible parties involved in the CAPA process. These notifications serve as unique reminders, ensuring that individuals are promptly informed about their tasks and impending deadlines. By doing so, the risk of missed actions or delays is considerably mitigated. This real-time communication not only keeps everyone on track but also supports a more responsive and accountable approach to handling corrective and preventive actions.
Reporting & Trend Analysis. Digital CAPA management tools also empower organizations to generate customized reports and analyze data trends. These serve as a window into the performance of the detection, rectification and thorough review of outliers within CAPA processes, providing invaluable insights for informed decision-making and continuous improvement initiatives.
In essence, the transformative power of digital tools is instigating a monumental shift in technology adoption, heralding a new era in establishing efficacious root cause analysis for successful CAPA management. By harnessing these tools, organizations can unlock avenues for enhanced efficiency, elevated quality standards and fortified regulatory compliance. This paradigm shift is paving a path toward sustained progress, where proactive problem-solving becomes the cornerstone of organizational growth and resilience.







