This year, more food manufacturing companies than ever are actively seeking software solutions for food safety and quality management. The majority of food businesses still collect food safety records using pen and paper, which is a time-consuming process and is far from reliable. Fortunately, food safety software is drastically changing the way HACCP and other QA/QC data becomes collected and stored. The following are some reasons food safety software is a must-have in 2016.
1. Overwhelming Amount of Paperwork
Many employees at food manufacturing facilities—whether meats, baked goods, or beverages—still rely on pen and paper forms, checklists, and log books to manage their food safety operations. This allows operator errors and omissions to happen far too easily. Even well managed systems that use paperwork can reach a tipping point. Quality managers already have a big enough workload, and piling on the job of verifying all paperwork only increases the chances of a failure.
2. Constant Change in Food Safety Standards
FSMA, the most sweeping reform of food safety laws in more than 70 years, was signed into law in 2011. While most food manufacturers have only been mandated to register, the time of more strict enforcement is coming. Global standards for food safety such as, SQF and BRC, fall within this generation of compliance. The necessities of these programs are constantly changing, becoming stricter and introducing new categories of requirements. Food safety software offers an effective way to deal with the ever-shifting landscape by providing evolving solutions managed by a software provider.
3. Consumer Confidence
Consumer expect a safe and healthy product, and as such, a company must instill a feeling of confidence. When a product is expected to provide nutrition, energy and enjoyment any incidence of foodborne illness will only prevent future confidence in the product. In addition, the consumer is likely to spread the news among peers about how he or she became sick. It is the duty of the food company to do everything in its power to process, store and prepare food in the safest way possible. That process starts with ensuring a higher level of reliability in a food safety program, which can be achieved by using food safety software.
4. Economics
No food company wants to deal with the hassle of a lawsuit from an outbreak or recall. These events can cost thousands, if not millions, of dollars in damages and cause serious loss to a company of any size. Although many companies carry insurance for these occurrences, there still is the loss of sale from consumer mistrust. Investing in software can help companies increase profits by improving efficiencies. For example, companies can measure these efficiencies not only from labor savings but also from significant savings in paper and toner (i.e., saving $2000–$5000 a year).
5. Operator Error and Omission
In an age in which documentation is becoming digitalized, food safety requirements are becoming more tedious. Even the smallest employee error can have massive repercussions. The cost of added staff is one thing, but the reliability of the data and adherence to schedule is another. Relying on inaccurate data collection may result in a recall or damage to a company’s brand.
Pencil whipping (faking paperwork) occurs for a variety of reasons, from employees taking short cuts to avoidance of writing down out-of-spec data. Manual record keeping has proven over time to be prone to errors. Employees, bored with the distraction of measuring and writing down dough or batch temperatures, piece weights or metal detector tests, and fill out forms with moot numbers just to complete the form.
6. Monitoring and Notification
Being aware of control points is another way in which companies can avoid disaster. Food safety software can give companies the ability to monitor oven and freezer temperatures, metal detectors, tests or any other control point in real time. This capability also alerts users when a control point is out of deviation (doing so at a glance), along with sending custom notifications, allowing a plant to quickly address problems while simultaneously properly documenting the issue.
7. Audits: Go from Stressful to Easy
An upcoming audit can be stressful for any company, involving numerous people who are gathering an abundance of documentation. With food safety software there is no reason to scramble to get documentation together or waste precious time preparing it. Auditors can simply view a company’s software for any requested documentation. For example, a company can produce random temperature logs, metal detector times, SSOPs, customer complaints and a variety of other documentation in just a few clicks.
8. The Technology Is Available
One of the reasons why so many companies continue to use paper and Excel-based systems today is because they are unaware of the abilities and functionality available to them. After completing the formidable task of attaining a new-found level of compliance, some companies may find it daunting to continue to go to the next step of converting to a computerized system. Finding user-friendly food safety software that has good customer support, as well as solutions that are customized to user needs, is not necessarily easy. The key is to find software and a supplier that can provide the right solution for your company and food safety program, whether it’s HACCP, BRC or SQF, and ensure that it fits within an acceptable budget. Consider not only the initial cost but also the cost of implementation. What resources will be required? How much can the supplier help with implementation? Having the right answers and the true costs will assist you in arriving at the correct solution.
If my company is GFSI-certified, is it also FSMA compliant? The answer is: With shared goals of producing safe food, coordinating preventive measures and ensuring continuous improvement, if your company is FSSC 22000 certified, you’re well on the road to FSMA compliance, according to Jacqueline Southee, Ph.D., U.S. Liaison, FSSC 22000. Southee discussed several areas in which FSSC 22000 aligns with FSMA as part of a recent Leadership Series, “GFSI in the Age of FSMA”.
Supply Chain Visibility
FSSC 22000 is applicable to all aspects of the supply chain and requires interactive communication (all of which must be documented), from the downstream level in ensuring raw materials and suppliers meet requirements of ISO 22000 framework to communication with customers and suppliers to verify and control hazards.
FSMA controls the hazard of food within the United States, says Southee, whereas GFSI certification is a global initiative, thereby extending supply chain visibility to foreign suppliers.
The Food Safety Plan
There has been much discussion surrounding building a FSMA-ready food safety plan and the migration from HACCP to HARPC. “HARPC can be referred to as HACCP with preventive controls,” says Southee. FSSC 22000 provides a flexible yet robust approach in a framework that is applicable to all situations (i.e., different manufacturers have different issues, such as producing ice cream versus baked goods). Rather than being prescriptive, the prerequisite program has the flexibility to apply to a particular situation. In addition, validation, verification, monitoring and documentation are an inherent part of the ISO 22000 approach and the FSSC 22000 certification.
FSSC 22000 serves as an effective tool in preparing companies for FSMA compliance. “We’re not a regulatory system; FDA has that domain,” says Southee. “They’re the ones that carry the responsibility of meeting those regulations. We work with everyone…to do the best job we can.”
Audit Readiness
Being audit ready all the time is a key part of preparing for FSMA. FSSC 22000 certifies a food safety management system (a three-year certification cycle) and requires internal audits of company performance, along with helping companies ensure that their records are organized at all times. The goal is to install a management system that enables constant monitoring, reevaluation and assessment as part of an ongoing process of keeping food safe, according to Southee. “If you’re certified and have an effective ongoing management system, unannounced audits won’t be an issue,” she says.
Food Safety Culture
FSSC 22000 and ISO 22000 provide a strong foundation for building food safety culture. ISO 22000 requires proof of management commitment to the food safety process, along with accountability, and for management to make resources available to see the food safety process through. “We agree that culture has to come from the top,” says Southee. “The personnel have to see that management is committed, and the culture will come from that commitment.” It also requires constant communication, up and down the supply chain as well as internally. This includes involving all employees and making sure that they know what they’re doing (i.e., training). “Everyone needs to know they’re valued and important, and how their function contributes to the function of safe food,” says Southee.
FSMA Alignment and Gap Analysis
There are sure to be some gaps when it comes to FSSC 22000 and FSMA. FSSC 22000 has commissioned a gap analysis to compare the preventive controls for human and animal food rules with the GFSI scheme and will add addendums as needed. Areas of review include a requirement to include food fraud into the hazard analysis and a review of unannounced audit protocol.
After much anticipation, FDA has finally published the FSMA final rules. If you’ve had time to dig into the details, you most likely noted the new initiative that requires companies to measure food safety culture. The industry is also seeing SQF, BRC and other GFSI audit schemes ramping up discussions around measuring food safety culture. However, FDA and GFSI audits aside, how do you create a culture for sustained compliance with this initiative? Follow these 10 tips to ensure your food safety culture is constant and in line with the new requirements
Set clear expectations for employees across the board. Photo credit: Dennis Burnett for Alchemy Systems
1: Create a solid foundation of programs, procedures and policies
Have a preset annual schedule for review and update of all programs, procedures and policies. Don’t let the schedule slide because there are competing priorities. A small pebble is all it takes to start ripple effect in the company, making it difficult to recover.
2: Set clear expectations, driven from the top down
Everyone should follow the rules and guidelines—from visitors to the CEO to the plant manager to the hourly employee. A “no exceptions” policy will drive a culture that is sustainable and drive a “this-is-just-how-we-do-things” mindset.
3: Use record keeping to ensure that food safety culture is well documented and data-driven
Collect the data that is measureable and non-subjective to help drive continuous improvement. If you collect it, you must do something with it. Good documentation is imperative to proving you did what you said you were going to do, especially in the event of an audit. Be stringent in training, and review all documentation before it hits the file cabinet to ensure it is accurate and appropriate.
4: Implement a robust continuous improvement process
Forward momentum through a continuous improvement process cannot be achieved unless management nurtures the program. If you are not continuously improving, you are falling behind.
5: Have a 360-degree approach to employee engagement with 24/7 awareness and communication
Top-down communication is critical to highlighting the priorities and needs of an organization and will not be effective unless an organized program is in place. Organizations that are not making the necessary pivots to communicate with the multiple generations within their workplace today will struggle to sustain change.
6: Foster an atmosphere of mutual respect
Treat people as you would like to be treated, turn the other cheek, etc. There may be lots of adages you quote, but which one best describes your facility and the relationships with management and peers on a daily basis?
7: Be sure employees have consumer awareness for the products they produce
Do your employees know who the end consumer is of the product that they are producing every day? Does your culture include a review of consumer complaints and customer complaints with your frontline workers? Listening in to a call center is a very powerful way to help employees understand what affects consumers and how their job is critical to avoiding a food safety or quality issue.
8: Create accountability across the board
Hold folks who do not support the culture in which you are striving to develop or maintain accountable, regardless of their position or stature.
9: Provide positive reinforcement. It’s the best motivator
Work to catch people doing things right and make a big fuss when you do. Positive reinforcement for a job well done is the most powerful motivator. It helps keep every team member on board with food safety commitments.
10: Celebrate often
We spend too much time at work not to celebrate all the good things that are accomplished. Whether it’s a cake and recognition for those that served in the armed forces on Veterans Day or a successful launch of a new product—celebrations are a great way to recognize and reinforce your employees’ hard work. Identifying and correcting mistakes should also be celebrated; they are fertile ground for making changes and provide great nutrients for continuous improvement.
As a result of the finalization of FSMA regulations on September 10, 2015, increasing requirements for procedures, documentation and testing will soon be impacting the food industry. The major effects on the food microbiology testing market will come in the form of an increase in the volume of samples that must be processed in accordance with the new FSMA rules, along with an improved emphasis on accurate and complete record keeping. The goals of FSMA are to create a new safety standard across the entire food chain. Increasing food pathogen testing will minimize possible recalls and the probability that dangerous food outbreaks occur.
Food manufacturers’ testing labs and third-party accredited testing labs can meet the demand for increased testing and improved record keeping in one of two ways: Via facility expansion or via implementing new technologies into the laboratory. While facility expansion might be an ideal long-term solution, it will not address the immediate surge in lab demand brought on by the new FSMA requirements, as it takes time to build new laboratories and hire employees. Implementing new technologies in the lab, then, makes the most sense, and where automation can be introduced into traditionally-manual processes, higher throughput may be realized using existing personnel and facilities. Automation further removes human error and improves the quality of the test being performed. The challenge for lab managers will be to objectively look at the current production bottlenecks in their testing operations and determine where technology may be introduced to increase throughput.
In addition to mandating additional testing, the FSMA regulations will require improved lab record keeping, as well as a new accreditation process that FDA will implement. The food testing industry faces the same dilemma that the healthcare industry faced some years ago in migrating from manual files to electronic health records. Lab notebooks have a real purpose in the lab, but their purpose should be more as a backup system to information that is gathered and stored electronically. While Laboratory Information Management Systems (LIMS) have been around for many years, their full potential in pathogen testing has yet to be realized. A properly designed LIMS provides an electronic database that not only aids in the accreditation process, but also allows samples to be traced throughout the testing facility. This allows positive test results to be screened from false positives or false negatives, and points to which equipment or procedures in the testing process need to be improved upon. LIMS technology for recording digital information can also trace user, operation time and performance specifications more accurately than lab notebook-based processes.
In summary, many changes are coming to the food industry as a result of increased regulations, presenting exciting opportunities to develop new products and technologies to alleviate the pain points within testing labs. The industry of food pathogen testing must change alongside the regulatory atmosphere in order to be competitive in a post-FSMA era.
SQF certification is an ongoing process; once you have attained it, you have effectively committed to a continuous improvement program for your business. Conducting regular, thorough and transparent management reviews will play a key role in ensuring that you continue to achieve your SQF goals.
The first and most important thing to understand about management reviews is that they don’t have to be a cost to your organization. In fact, when conducted correctly, these reviews should become the cornerstone of your continuous improvement program, assisting you in measuring company performance against documented objectives.
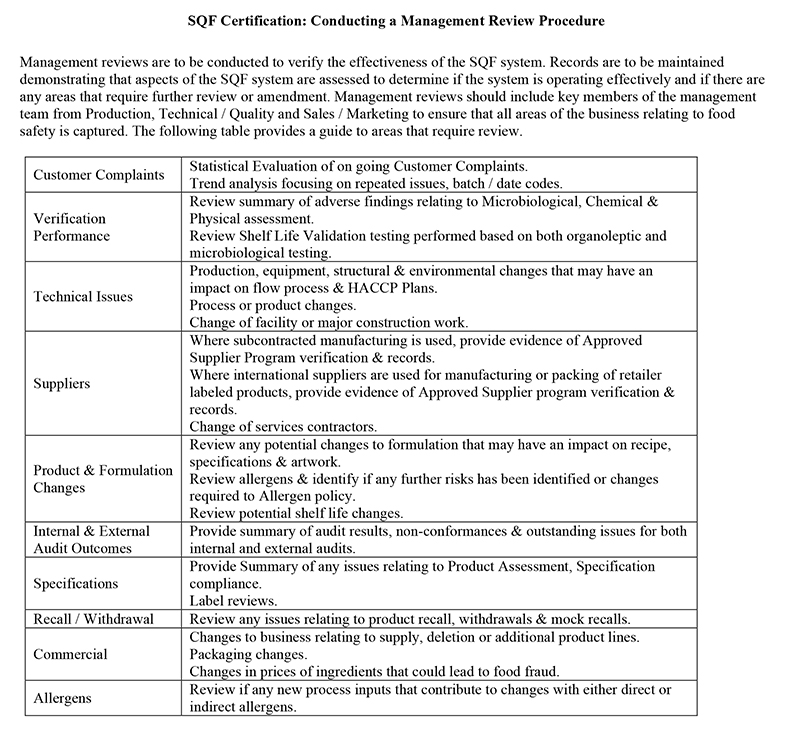
Once you’ve committed to a management review, it’s important to be aware of the fact that these reviews take significant preparation. The senior management team will be responsible for reviewing the SQF system, preparing for and documenting the review procedure in its entirety. As outlined in clause 2.1.2.4 of the SQF system, there are four key elements against which company performance must be reviewed:
The SQF policy manual
Internal and external audit findings
Corrective actions (investigation and resolution)
Customer complaints (investigation and resolution)
Click to enlarge this document on how to conduct a Management Review Procedure
The management team is required to make adequate resources available for food safety and quality objectives and to support the development, implementation and maintenance and ongoing improvement of the SQF system (clause 2.1.2.3). This team must also establish processes to improve the effectiveness of the SQF system in order to demonstrate continuous improvement (clause 2.1.2.9).
Keeping Good Records
One cannot overstate the importance of thorough and effective documentation of the review, so remember the mantra: If you don’t write it down, it didn’t happen.
Every aspect of your review must be clearly and thoroughly documented. These records are important documents in the SQF audit process and also as benchmarks against which company performance and subsequent reviews can be measured.
Management Review Process
The management review process comprises four key stages:
Identifying objectives
Collecting and analyzing data
Management review meeting
Disseminating results
1. Identifying Objectives
Identifying objectives should be your first step. You cannot start collating data before you know what you’ll be measuring it against. Objectives are essentially short statements that outline what you want your company to achieve in specific business areas. They should encompass a whole-of-company approach in that they should contribute to overarching company goals rather than delivering a one-sided or fragmented approach to continuous improvement. It is important to note that objectives are neither mission statements nor warm and fuzzy statements.
As a general rule of thumb, objectives should be:
Measurable. They should outline specific quantities and quotas for what you are trying to achieve.
Business-specific. Objectives should be relevant to the future of your business and to the individual business improvement process. Don’t worry about what your competitors are doing.
Relevant. Make sure the objectives will have a significant and lasting impact on your business once they are achieved. This might mean choosing complex objectives versus taking the easy way out.
Linked to a key performance indicator. Every objective should contribute to the company’s KPIs and be part of its broader strategic goals..
Clearly defined. Be clear about what you are trying to achieve and how you plan to achieve it.
Documented. Write your objectives down! The review process, future audits and your ongoing improvement efforts will be measured against them, so it is important to accurately capture the details.
Communicated across the breadth of the organization. Don’t make the mistake of keeping them in a filing cabinet somewhere. Put them in the lunchroom, put them in induction packs, hand them out during annual updates—share them with your team so that everybody understands the goals.
2. Collecting and Analyzing Data
Data collection is one of the most time consuming components of the management review process, so have a clear game plan ready before starting. Ask the following questions:
a. What information must be extracted from company records?
Determining the sort of data you need depends largely on your management review objectives. Your data should deliver insights on company performance and provide an evidence base for changes you make to your business processes. As a food company, your SQF systems should monitor and collect data on a wide range of business activities, including:
Swab and microbiological results from cleaning processes
Microbiological results from water supply and product testing, or raw material testing
Insurance and credit claims
Chemical product results
Mock recall tests
Product traceability tests
Think about the data sources that respond directly to the objectives you developed for your review. You should be leveraging the systems you already have to collate information, with a view to feeding the information back into work practices to create a loop of continuous improvement.
b. What format is the data in and how can you make it accessible?
Like most food companies, your company is likely to be home to a mix of paper-based and electronic data. Different data types throw up different challenges when it comes to data acquisition. Paper forms are easy for staff to use and easy for management to collect, but they require double handling in order to translate relevant data into a format that can be analyzed with any degree of depth. Electronic data collection is more useful when it comes to number crunching, but you need access to specific expertise when it comes to data extraction if you want your data to work effectively for you. Newer, more sophisticated data management systems will help automate data collection activities and will also ensure that your database systems can communicate with each other to share relevant information.
Data access is another issue to think about early in the process. Who is responsible for all the different data sets that must be accessed for the review? Is he or she the right person to manage that data? Will he or she be available at the time you need to access the data before the review?
c. Who will analyze the data, and do you have the requisite skills in-house?
Another important element of the pre-planning process is the question of who will analyze the data once it has been extracted from your systems. Does your staff have the skills and time required to conduct effective statistical analysis? Do you have suitable software systems to support the analysis process? If so, you’ll need to allocate each data analysis task to a staff member, making roles, responsibilities and timelines for the analysis process clear. In addition, ensure that all managers have access to (and an understanding of) the previously identified management review objectives so they can target their analysis appropriately.
If you don’t have the requisite data management skills in-house, you need to think through potential solutions during the pre-planning stage, whether it involves providing training for staff or bringing in outside expertise.
Prior to the data analysis process, you should also be looking at your raw data and reviewing data outliers (that is, data that lies above or below the mode)—these can sometimes provide interesting insights about your processes and procedures. It’s important to have an experienced quality assurance team on board before you start assessing your outliers; your QA team should already understand the variables within your processes and procedures, and will therefore be able to identify whether the outliers are one-off data spikes or trends requiring further investigation.
3. Management Review Meeting
a. Meeting preparation
Meticulous preparation is key to a successful management review meeting. Ensuring all meeting attendees are on board with the agenda and meeting outcomes is a good way to get started. First, develop a written agenda and timeline for the review. Think about the timing of your meeting in conjunction with your unannounced SQF audit. Set the date of the meeting with enough time to ensure close off of the corrective actions issued as a result of the meeting. Yes, if you’re doing it properly, you should have corrective actions issued.
Next, you’ll need to get all participating staff members up to speed with the review process. Pull together the requirements of the review, the relevant SQF clauses, customer requirements and company objectives, and put them into a document that can be circulated well in advance of the meeting. Identify the individual managers who will be involved with the review—that is, the managers whose business areas will make a contribution to, or be impacted by, the review process itself. Ask participating managers to prepare the data that is relevant to their key performance areas and to the management review objectives you’ve identified, as this will be distributed at the meeting.
b. At the meeting
Good record keeping is particularly important during the management review meeting itself. Poor documentation can undermine the review and its subsequent outcomes, not to mention the SQF auditing process. As such, it’s important to capture the details of the discussion that occurs at the meeting—you need more than just a few scribbled notes. Nominate a scribe whose sole responsibility is to take notes at the meeting. Ensure they capture (at a minimum) the following:
Meeting date and time
Meeting attendees and no-shows
Discussion of each agenda topic
Key items
Outcomes
Conclusions
Action points
Objectives. Were they met? If not, why not? Do they need to be modified for next year? If so, why, and to what?
Action points, role responsibilities and verification of actions being undertaken
Conclusions
The scribe should also be responsible for compiling all supporting documentation from the meeting for audit purposes and for review prior to future management review meetings.
4. Disseminating Results
Once the review process is completed, any processes and procedures identified as requiring modification must be updated. This is also the time to follow up on action items—your meeting documentation should clearly show who is responsible for each item and their respective tasks. Engage with the staff members responsible for each item at regular intervals to chart their progress, and follow up again at subsequent management meetings to ensure that all actions have been completed.
Communicating review outcomes is very important. Charting a specific course requires an understanding of, and commitment to, that course by everyone involved in the navigation process—and you need your team to understand where you’re going so that they can help you get there. This means communicating all the outcomes, both good and bad, to all relevant personnel, and taking the time to thank and congratulate staff who were instrumental in achieving positive results.
The last step of the review process is to share your success—don’t let your hard work go unnoticed! Communicate the value of the review both to your team and your superiors, and demonstrate the link between the review itself and the achievement of individual, team and company KPIs.
Conclusion
A completed management review will deliver significant benefit to your company in the form of documented, measurable and communicated objectives that are reviewed on an annual basis. Additional benefits include:
Increased business value
Enhanced safety (for staff) and quality of product (for clients)
A more targeted approach to business strategy
Reduced financial risk and increased profitability
In order to ensure that a food testing laboratory maintains a quality management system that effectively manages all aspects of laboratory operations that affect quality, there are numerous records, reports and data that must be recorded, documented and managed.
Gathering, organizing and controlling all the data that is generated, managed and stored by food testing laboratories can be challenging to say the least. As the ISO Standards and regulatory requirements for food testing laboratories evolve, so does the need for improved quality data management systems. Historical systems that were very efficient and effective 10 years ago, may no longer meet the demanding requirements for ISO 17025 certification. One way to meet the challenge is to turn to automated solutions that eliminate many of the mundane tasks that utilize valuable resources.
There are many reasons for laboratories to seek this certification, including to enhance reputation, gain a competitive advantage, reduce operational costs, and meet regulatory compliance goals. A major advantage for food testing laboratories to obtain ISO 17025 Certification is that is tells prospective clients that the laboratory has a strong commitment to quality, and they hold the certification to prove it. This certification not only boosts a laboratory’s reputation, but it also demonstrates an organization’s commitment to quality, operational efficiency and management practices. Proof of ISO 17025 Certification eliminates the need for independent supplier audits, because the quality, capability and expertise of the laboratory have been verified by external auditors. Many ISO Certified laboratories will only buy products (raw materials, supplies and software) and services from other ISO-certified firms so that they do not need to do additional work in qualifying the vendor or the products.
There are many areas in which a LIMS supports and promotes ISO 17025 compliance. Laboratories are required to manage and maintain SOPs (standard operating procedures) that accurately reflect all phases of current laboratory activities such as assessing data integrity, taking corrective actions, handling customer complaints, managing all test methods, and managing all documents pertaining to quality. In addition, all contact with clients and their testing instructions should be recorded and kept with the job/project documentation for access by the staff performing the tests/calibrations. With a computerized LIMS, laboratory staff can scan in all paper forms that arrive with the samples (special instructions, chain of custody (CoC), or any other documentation). This can be linked to the work order and is easy assessable by anyone who has the appropriate permissions. The LIMS provides extensive options for tracking and maintaining all correspondence, the ability to attach electronic files, scanned documents, create locked PDFs of final reports, COAs (Certificate of Analysis), and CoCs.
Sample Handling and Acceptance
Laboratories are required to have a procedure that defines all processes that a sample is subjected to while in the possession of the laboratory. Some of these procedures will relate to sample preservation, holding time requirements, and the type of container in which the sample is collected or stored. Other information that must be tracked includes sample identification and receipt procedures, along with acceptance or rejection criteria at log-in. Sample log-in begins and defines the entire analysis and disposal process, therefore it is important that all sample storage, tracking and shipping receipts as well as sample transmittal forms (CoC) are stored, managed and maintained throughout the sample’s analysis to final disposal. To summarize, the laboratory should have written procedures around the following related to sample preservation:
Preservation
Sample identification
Sample acceptance conditions
Holding timesShipping informationStorage
Results and Reporting
Disposal
The LIMS must allow capture and tracking of data throughout the sample’s active lifetime. In addition, laboratories are also required to document, manage and maintain essential information associated with the analytical analysis, such as incubator and refrigerator temperature charts, and instrument run files/logs. Also important is capturing data from any log books, which would include the unique sample identifier, and the date and time of the analysis, along with if the holding time is 72 hours or less or when time critical steps are included in the analysis, such as sample preparations, extractions, or incubations. Capturing the temperature data can be automated such that the data can be directly imported into the LIMS. If there is an issue with the temperature falling outside of a range, an email can automatically be spawned or a message sent to a cell phone to alert the responsible party. Automation saves time and money, and can prevent many potential problems via the LIMS ability to import and act on real-time data.
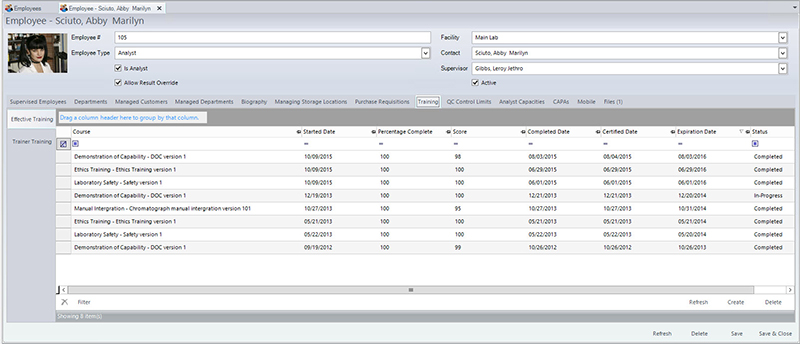
If any instrumentation is used in the analysis, the following information must also be recorded in the instrument identification (to ensure that it is in calibration, and all maintenance and calibration records are current), operating conditions/parameters, analysis type, any calculations, and analyst identification. In addition to analyst identification, laboratories must also keep track of analyst training as it relates to their laboratory functions. For example, if an analyst has not been trained on a particular method or if their certification has expired, the LIMS will not allow them to enter any result into the LIMS for the method(s) that they have not been trained/certified to perform. The LIMS can also send automated alerts when the training is about to expire. Figure 1 shows a screen in the LIMS that manages training completed, scheduled, tests scores, and expiration dates of the training, along with the ability to attach any training certificates, exams, or any other relevant documentation. Laboratory managers can also leverage the LIMS to pull reports that compare analyst work quality via an audit report. If they determine that one analyst has a significant amount of samples that require auditing, they can then investigate if there is a possible training issue. Having immediate access to data allows managers to more rapidly identify and mitigate potential problems.
LIMS manages a variety of aspects in training, including when it has been completed, scheduled, tests scores, and expiration dates. (Click to enlarge)
Another major area that a LIMS can provide significant benefit is around data integrity. There are four main elements of data integrity:
Documentation in the quality management system that defines the data integrity procedure, which is approved (signed/dated) by senior management.
Data integrity training for the entire laboratory. Ensures that the database is secure and locked and operates under referential integrity.
Detailed, regular monitoring of data integrity. Includes reviewing the audit trail reports and analyzing logs for any suspicious behavior on the system.
Signed data integrity documentation for all laboratory employees indicating that they have read and understand the processes and procedures that have been defined.
The LIMS will enhance the ability to track and manage data integrity training (along with all training). The LIMS will provide a definition of the training, the date, time, and topic (description); instructor(s); timeframe in which the training is relevant, reminders on when it needs to be repeated; along with certifications, quiz scores, copies of quizzes, and more. With many tasks, the LIMS can provide managers with automated reports that are sent out at regular time intervals, schedule training for specific staff, provide them with automatic notification, schedule data integrity audits, and to facilitate FDA’s CFR 21 part 11 compliance (electronic signatures). The LIMS can also be configured to automatically have reports signed and delivered via fax or email, or to a web server. The LIMS manages permissions and privileges to all staff members that require access to specific data and have the ability to access that data, along with providing a secure document control mechanism.
Laboratories are also required to maintain SOPs that accurately reflect all phases of current laboratory operations such as assessing data integrity test methods, corrective actions and handling customer complaints. Most commercial LIMS provide the ability to link SOPs to the analytical methods such that analysts can pull down the SOP as they are doing the procedure to help ensure that no steps are omitted. Having the SOPs online ensures that everyone is using the same version of the locked SOPs, which are readily available and secure.
Administrative Records, Demonstration of Capability
Laboratories are required to manage and maintain the following information on an analyst working in the laboratory: Personal qualifications and experience and training records (degree certificates, CV’s), along with records of demonstration of capability for each analyst and a list of names (along with initials and signatures) for all staff that hold the responsibility to sign or initial any laboratory record. Most commercial LIMS will easily and securely track and manage all the required personnel records. Individuals responsible for signing off on laboratory records can be configured in the LIMS to not only document the assignment of responsibility but also to enforce it.
Reference Standards and Materials
Because the references and standards that laboratories use in their analytical measurements affect the correctness of the result, laboratories must have a system and procedures to manage and track the calibration of their reference standards. Documentation that calibration standards were calibrated by a body that can prove traceability must be provided. Although most standards are purchased from companies that specialize in the creation of reference standards, there are some standards that laboratories create internally that can also be traced and tracked in the LIMS. Most commercial LIMS will also allow for the creation, receipt, tracking, and management of all supplies in an inventory module, such that they document the reference material identification, lot numbers, expiration date, supplier, and vendor, and link the standard to all tests to which it was linked.
The ISO 17025 Standard identifies the high technical competence and management system requirements that guarantee your test results and calibrations are consistently accurate. The LIMS securely manages and maintains all the data that supports the Quality Management System.
Key advantages of food testing laboratories that have achieved ISO 17025 Certification with a computerized LIMS that securely and accurately stores all the pertinent data and information:
Proof of ISO 17025 Certification eliminates the need for supplier audits, because the quality, capability and expertise of the laboratory have been demonstrated by the certification.
Knowledge that there has been an evaluation of the staff, methods, instrumentation and equipment, calibration records and reporting to ensure test results are valid.
Verification of operational efficiency by external auditors that have validated the quality, capability and expertise of the laboratory.
Defines robust quality controls for the selection and authentication of methods, analyzing statistics, controlling and securing data.
Clearly defines each employee’s roles, responsibilities and accountability.
Confidence that the regulatory and safety requirements are effectively managed and met in a cost efficient-manner.
With nearly one in every six Americans falling prey to foodborne illnesses each year, food safety is a major public health issue. For several decades, current Good Manufacturing Practices (cGMPs) provided the basic food safety framework for manufacturers. However, these guidelines were not sufficient to cover all potential food safety hazards. In the 1960s, NASA asked Pillsbury to manufacture the first foods for space flights, and so the Hazard Analysis & Critical Control Point (HACCP) system was born. HACCP was later endorsed by the Codex Alimentarius Commission, which was formed by the Food and Agriculture Organization of the United Nations and the World Health Organization in 1963.
HACCP is a global standard and its principles are the defining elements of ISO 22000, BRC and SQF, all premiere global food safety standards. In 1996, an E. coli outbreak in Scotland claimed 10 lives. The Pennington report in the aftermath of this tragedy recommended use of HACCP by all food manufacturers to ensure food safety. While HACCP is mandatorily used for seafood, juice and USDA-regulated meat processing, it could not win universal acceptance across the food industry; most of the food industry sectors rely on cGMP for providing a food safety framework.
The number of people affected by foodborne illnesses can be attributed to a flawed food safety system. Thinking caps were put on and President Obama’s administration rigorously pursued what it hoped would be an effective food safety paradigm. On July 4, 2012 Hazard Analysis and Risk Based Prevention Control (HARPC) was introduced under FSMA section 103. Although the system is still a work in progress and FDA has yet not disclosed the regulations that will determine the functionality of HARPC, the agency is bound to issue the regulations by August 30, 2015. HARPC will become effective 60 days following this date, and companies will be required to enforce HARPC within a period of 12 to 36 months, depending on the size of a facility.
HARPC is designed along the lines of HACCP but is meant to be more comprehensive. For a “Simple Simon” it would be tough to differentiate between the two, but HARPC provides an all-encompassing food safety structure by focusing on preventive controls to make food safety more iron clad. With the exception of exempted facilities, HARPC will apply to all facilities subjected to FDA’s Bioterrorism Facility Establishment registration. All such facilities will be expected to enforce a functioning and adequate HARPC plan. Failure to do so and FDA would be authorized to take legal actions such as issuing a public warning letter or an import alert (in case of a foreign supplier), initiating criminal proceedings against a non-compliant facility, or suspending food facility registration of a facility until requirements are met. By doing so, FDA has put the onus squarely on the shoulders of respective facilities. Companies will be required to do a lot more and should expect deeper FDA involvement. Expert help to enforce a rather complex HARPC protocol seems unavoidable; there is a fair chance that users could find themselves lost in the translation and may end up facing FDA’s wrath if their plan is inadequate. Let me break it down a bit more and distinguish the main differences between HARPC and HACCP.
Qualified Food Safety Experience. HARPC requires one member of a company to be the qualified individual to complete an entire food safety plan. This means that said individual has undertaken education from a credible institution and gained experience by completing it. HACCP requires at least one person to be HACCP certified, but the plan must be constructed by a team of people.
Process Flow Diagram. Under the HACCP standard, food safety plans must include a clear flow diagram outlining the process, from start to finish, that the ingredients will take throughout your facility. HARPC has no regulations regarding this.
Hazard Variables. Traditionally, hazards were limited to biological, chemical and physical hazards under the HACCP paradigm. Yet, under HARPC, you must also outline Radiological and Terrorism hazards.
Controlling Hazards. Here is largely where the main difference lies: How to control a hazard. HACCP requires companies to mention their critical control points as well as outline a prerequisite program (PRP), although this has no set requirements. HARPC requires you to apply a sanitation preventive control to the hazards, which looks at monitoring, confirmation, corrective action, reviewing records and re-analyzing.
Reviewing the Plan. HACCP requires the individual in charge to review all HACCP documentation every year. This is in comparison to HARPC, which requires a facility to reanalyze its plan every three years.
Recall Plans. Recalls, as required under HARPC, are a special type of incident, with all of the attributes necessary to create and manage a recall plan. HACCP does not have such a requirement.
Use Software to Implement HARPC Plan
Using software can make life easier when it becomes time to implement a HARPC plan. Documentation is an important part of the HARPC system, and software can help generate most of the documents used to establish the plan. Such a system can link regulatory requirements with procedures and customize several aspects of the system during run time.
A risk analysis component of software helps a user identify the likelihood and severity of a particular hazard (a HARPC requirement). HARPC also requires sanitation control procedures at food surface contact points; software features can support cross contamination points to which hazards are assigned and controlled. Software also allows users to define equipment, with a facility to schedule and record calibration, maintenance, and verification activities, including management task assignment to satisfy HARPCs provisions regarding sanitation of utensils and equipment. In addition, it has the provision to document procedures as required by HARPC and can also flag employees for refresher training if they are involved in a violation.
Software also enables users to electronically record inspections, which satisfies the obligation under HARPC to carry out an environmental monitoring program (for pathogen controls). Interestingly, sensors could also be integrated with logging facilities to automatically collect sensor data, which could then be used to send out alerts if there is an abnormality. Software systems can also accommodate coverage of allergen hazards and run a food allergen control plan, including documentation of the process.
An incident management plan can assign and track corrective actions, root causes, employee retraining tasks, and preventive measures to individuals, and recall plans can be created and managed using the system. As many inspectors prefer remote review of documentation, software can provide such remote access, allowing inspectors to conduct off-site document reviews. This process can reduce on-site inspection times from five to three days. A list of approved suppliers can be maintained as well, and these suppliers can be linked to receiving functions, enabling users to receive and maintain a detailed and comprehensive record of ingredients.
HARPC is a reality that will have to be embraced very soon. Using software is a simple solution for the tough times that lay ahead for the food industry. It can serve as an all-encompassing and one-stop-shop for businesses that need help enforcing HARPC plans.
Although FDA immediately gained increased authority for records access upon the signing of FSMA in January 2011, some companies haven’t gotten the memo yet.
The following best practices will help food and beverage manufacturing facilities get inspection ready and offer guidance on how to push back if overreaching occurs.
Keep a paper trail. Documentation and recordkeeping is crucial. Disclosure is relevant only to the documents that FDA is allowed to have, such as production documentation. This does not apply to proprietary/trade secret information (i.e., financials and recipes).
Appoint a designated person to guide FDA during the inspection. This person should be the sole contact for all requests of documents, information, samples, etc., and accommodate requests for information from the inspector, as long as they are reasonable and in scope. This point person must have adequate training and experience in regulatory requirements and recent changes to have an awareness of what records inspectors are and are not technically authorized to review or access. If you are unsure of what the inspector is asking for at any point, we have a word of advice: Never speculate; always ask for clarification.
Duplicate everything. If the inspector takes swabs or photos, shadow him/her and take duplicate swabs and/or photos so you have a record of what was taken by the agency. It’s another question whether to take the swabs to test or not—but at least have a replica of what the agency has so you have the same or very similar information.
Engage legal if you feel that lines are being crossed. When you think inspectors have crossed the line into records to which they should not have access, it’s okay to respectfully state that you would like to speak with your legal counsel prior to disclosing the record. Make it clear you are not outright refusing to produce the document; rather that you are unsure what the company policy and/or regulation is on disclosure of that particular document/piece of information; and, as such, you wish to seek legal counsel prior to disclosing.
If you do refuse outright to produce requested information, do so wisely. Inspectors are given clear guidance on what actions to take if met with refusals. Criminal provisions of the Act may be invoked but this is in the most egregious of cases. Some of the guidance relevant to inspectors that food companies should be aware of are stated in the FDA Inspector Inspection Manual and are included below to give you an idea of the response you may receive to an outright inspection refusal or refusal to allow copying of records:
2.6 – INSPECTION WARRANT. A refusal to permit inspection or a refusal to permit access to or copying of records may invoke criminal provisions of sections 301(e) and 301(f) of the FD&C Act [21 U.S.C. 331(e), (f)].
However, as stated in 5.2.5, the request still needs to be fair and reasonable:
2.5 – INSPECTION REFUSAL. Refusal as used in your IOM means refusing to permit an inspection or prohibiting you from obtaining information to which FDA is entitled under the law. In the case of a refusal you must show your conduct was reasonable, fair, and you exercised reasonable precaution to avoid refusal. You must have shown your credentials and given the responsible individual a properly prepared and signed Notice of Inspection, FDA 482.
2.5.2 – Refusal to Permit Access to or Copying of Records. If management objects to the manner of the inspection or coverage of specific areas or processes, do not argue the matter but proceed with the inspection. However, if management refuses to permit access to or copying of any record to which you are entitled under law, call attention to Section 301(e) of the FD&C Act[21 U.S.C. 331] or applicable sections of the PHS Act. If management still refuses, proceed with the inspection until finished.
Mark documents confidential/proprietary. Mark all documents and materials disclosed to the inspector as Confidential/Proprietary or Company Confidential Information if you wish to even attempt to maintain confidentiality and keep the information from being disclosed in a Freedom of Information Act (FOIA) release. If not marked as such, 483 information is generally discoverable through FOIA requests.
Request your own FOIA. After the inspection, the FDA inspector will write a report called the Establishment Inspection Report (EIR). Best practice is for the company to submit a FOIA request for both a redacted (confidential information removed) and an unredacted copy of the EIR to ensure the information in the report matches the one that the inspector provided to the company. This is requested through the FDA Freedom of Information Office in Rockville, Maryland.
If you don’t agree with a 483 item, it can be disputed/appealed. This is done in the standard 483 response in which a firm provides information on corrective actions it will take on issues that do need correcting. As stated in the FDA Field Directive No. 120, “The firm may request clarification, criticize FDA 483 items, disagree with the FDA 483, or raise other questions or issues. In these cases, the District will evaluate the firm’s information and send the District’s conclusion to the firm. A copy shall also be sent to the official establishment file.”
The product recall at Blue Bell Creameries earlier this year is yet another example of food safety issues negatively impacting food marketers, growers, processers and manufacturers. We all remember the Peanut Corporation of America’s salmonella outbreak in 2008 and the Jensen Farms listeria outbreak in 2011. Salmonella-tainted eggs in 2010, E. coli in strawberries in 2011, and listeria in caramel apples last Halloween combined with dozens of others during the last six years, have sickened thousands and killed dozens of people.
The brand reputation impact from the incidents at Peanut Corporation of America and Jensen Farms was terminal—both companies went bankrupt. The effect on Blue Bell, while likely not fatal, is expected by industry experts to be substantial and include loss of revenue and market share. The company has already announced plans to lay off more than 1,000 workers as a result of the recall.
In addition, growers saw cantaloupe consumption take a nosedive after the Jensen Farms listeria outbreak, which was one of the worst foodborne illness outbreaks in U.S. history in terms of number of deaths. They are only now seeing sales levels return to those before the incident. And because the farm itself went out of business, personal injury lawyers went after the companies that sold the disease-ridden cantaloupes—the retailers. By virtue of last year’s out-of-court settlement by Walmart on the Jensen Farms lawsuit, both suppliers and retailers are now responsible for everything they sell.
Enter the Food Safety Modernization Act, signed in 2011 and about to begin finalization in August. FSMA mandates that retailers and suppliers have documentation that verifies their supply chain’s regulatory compliance is readily accessible for government inspection. Add these records to the business relationship records that retailers and suppliers should already be maintaining (including indemnifications and certificates of insurance that help manage brand risk), and you’d think our risk of foodborne illness is about be eradicated.
Although FSMA represents the most sweeping change to our food safety laws in the last 70 years, it may not have the greatest impact where the supply chain is most vulnerable. Today the largest suppliers that sell the majority of our food have very sophisticated systems to ensure safe food production and transportation. This group will have the easiest path to compliance with FSMA, and they most likely already hold themselves to a higher standard. It’s actually the smaller suppliers, which likely do not have the available resources or sophistication to comply with FSMA requirements, that will be exempt from certain documentation under FSMA based on their size. This group of suppliers is growing rapidly to meet consumer desire for fresh food that is locally grown and produced. Unfortunately for them, it’s only a matter of time before wholesalers and retailers decide that the risk is too great to continue to do business with these small suppliers.
The good news is that technology exists that can help small suppliers reduce risk in their extended supply chains. Affordable, interoperable systems have been developed to address the market need for receiving, storing, sharing and managing regulatory, audit and insurance documentation. Suppliers of any size can also track products as they move through the supply chain and trace them back in the event of a recall. This move to automation will help all suppliers not only meet the demands of FSMA, but also establish a base for retailer and consumer demands for transparency in the supply chain going forward.
Having a comprehensive food safety system is quickly becoming a competitive advantage. Retailers and consumers are looking for those suppliers that have an unblemished safety record and are transparent about their safety processes, so the time is now for small suppliers to hold themselves to a higher standard than FSMA requires for future business opportunities. The stakes are just too high for retailers and wholesalers to not verify that everything they sell to consumers is produced and transported safely.
Gary Nowacki, CEO of TraceGains, highlights the concerns that came out of an Ask the Expert discussion about Supplier Qualifications and Management at the 2014 Food Safety Consortium.
“A common takeaway–whether it was small, mid-size or large companies–was the frustration of having to do more audits, and the growing demand for more paperwork. A question that was often asked was that ‘my customer is asking me to subscribe to a different audit, and that defeats the promise of GFSI, that it would lead to fewer audits.’ My advice to them is to not just blindly agree, but ask the customer politely what exactly they are looking for, and see if they can address that. Another frustration related to the increase in paperwork and the time and resources consumed in filling these plethora of forms. So there was a discussion about how we can standardize these. And people are looking to get automated solutions as they are not getting more headcount.”
This website uses cookies so that we can provide you with the best user experience possible. Cookie information is stored in your browser and performs functions such as recognising you when you return to our website and helping our team to understand which sections of the website you find most interesting and useful.
Strictly Necessary Cookies
Strictly Necessary Cookies should be enabled at all times so that we can save your preferences for these cookie settings.
We use tracking pixels that set your arrival time at our website, this is used as part of our anti-spam and security measures. Disabling this tracking pixel would disable some of our security measures, and is therefore considered necessary for the safe operation of the website. This tracking pixel is cleared from your system when you delete files in your history.
We also use cookies to store your preferences regarding the setting of 3rd Party Cookies.
If you visit and/or use the FST Training Calendar, cookies are used to store your search terms, and keep track of which records you have seen already. Without these cookies, the Training Calendar would not work.
If you disable this cookie, we will not be able to save your preferences. This means that every time you visit this website you will need to enable or disable cookies again.
Cookie Policy
A browser cookie is a small piece of data that is stored on your device to help websites and mobile apps remember things about you. Other technologies, including Web storage and identifiers associated with your device, may be used for similar purposes. In this policy, we say “cookies” to discuss all of these technologies.
Our Privacy Policy explains how we collect and use information from and about you when you use This website and certain other Innovative Publishing Co LLC services. This policy explains more about how we use cookies and your related choices.
How We Use Cookies
Data generated from cookies and other behavioral tracking technology is not made available to any outside parties, and is only used in the aggregate to make editorial decisions for the websites. Most browsers are initially set up to accept cookies, but you can reset your browser to refuse all cookies or to indicate when a cookie is being sent by visiting this Cookies Policy page. If your cookies are disabled in the browser, neither the tracking cookie nor the preference cookie is set, and you are in effect opted-out.
In other cases, our advertisers request to use third-party tracking to verify our ad delivery, or to remarket their products and/or services to you on other websites. You may opt-out of these tracking pixels by adjusting the Do Not Track settings in your browser, or by visiting the Network Advertising Initiative Opt Out page.
You have control over whether, how, and when cookies and other tracking technologies are installed on your devices. Although each browser is different, most browsers enable their users to access and edit their cookie preferences in their browser settings. The rejection or disabling of some cookies may impact certain features of the site or to cause some of the website’s services not to function properly.
Individuals may opt-out of 3rd Party Cookies used on IPC websites by adjusting your cookie preferences through this Cookie Preferences tool, or by setting web browser settings to refuse cookies and similar tracking mechanisms. Please note that web browsers operate using different identifiers. As such, you must adjust your settings in each web browser and for each computer or device on which you would like to opt-out on. Further, if you simply delete your cookies, you will need to remove cookies from your device after every visit to the websites. You may download a browser plugin that will help you maintain your opt-out choices by visiting www.aboutads.info/pmc. You may block cookies entirely by disabling cookie use in your browser or by setting your browser to ask for your permission before setting a cookie. Blocking cookies entirely may cause some websites to work incorrectly or less effectively.
The use of online tracking mechanisms by third parties is subject to those third parties’ own privacy policies, and not this Policy. If you prefer to prevent third parties from setting and accessing cookies on your computer, you may set your browser to block all cookies. Additionally, you may remove yourself from the targeted advertising of companies within the Network Advertising Initiative by opting out here, or of companies participating in the Digital Advertising Alliance program by opting out here.