Find records of fraud such as those discussed in this column and more in the Food Fraud Database. Image credit: Susanne Kuehne.
In the United States, the FDA is responsible for regulations and recommendations to protect public health, which includes the prevention of any type of food adulteration (unintentional contamination, intentional adulteration, and food fraud – or “economically motivated adulteration”). FSMA (the Food Safety Modernization Act) resulted in new regulations and guidance with strategies to reduce all types of risks in food facilities. It was the most comprehensive reform of FDA’s food safety regulations in more than 70 years.
By Cori Goldberg, Adam Brownrout, John Kendzior No Comments
On October 29, 2019, the USDA released its long-awaited draft rule establishing a domestic hemp production plan, providing clarity to growers and ancillary businesses about how the USDA will regulate the hemp crop. The USDA, under authority provided by the 2018 Agricultural Improvement Act (2018 Farm Bill), was tasked with promulgating regulations and guidelines to establish and administer a program for the production of hemp in the United States. This rule has now arrived and been published in the Federal Register. The rule provides requirements for all state and tribal hemp production plans including requirements for testing hemp, licensing growers, disposing of non-compliant hemp, and collecting and storing information related to hemp production. The USDA will now accept public comment on the rule until December 30, 2019.
Although the USDA rule will greatly contribute to the expansion of legally grown hemp in the United States, this rule does not alter the law regarding CBD foods and CBD dietary supplement products. This is because the 2018 Farm Bill left intact FDA’s authority to regulate the sale and marketing of CBD foods, dietary supplements, drugs, and cosmetics, as those product types fall under FDA’s purview generally. FDA has allowed the sale of CBD cosmetics, with certain restrictions, and companies may submit CBD products to FDA through FDA’s drug approval process. However, it has maintained that the addition of CBD to foods and dietary supplements is illegal. Under the federal Food, Drug, and Cosmetic Act (FDCA), once a substance is approved as an Active Pharmaceutical Ingredient (API) in an FDA-approved drug, that substance may not be placed into interstate commerce in a food. Also under the FDCA, once a substance is approved as an API in an FDA-approved drug, that substance is excluded from the definition of a dietary supplement. FDA approved the pediatric epilepsy drug, Epidiolex, whose API is CBD. Therefore, FDA has concluded that CBD may not be placed into foods in interstate commerce and that CBD products are excluded from the dietary supplement definition and therefore may not be sold as dietary supplements. The USDA rule does nothing to change the legal status of CBD food or dietary supplement products. Thus, despite the expected increase of hemp availability following the passage of the USDA rule, CBD companies must wait for the FDA green-light in order to manufacture or sell hemp-derived CBD food products lawfully.Learn more about important regulatory & quality issues in the cannabis space from Cannabis Industry Journal
However, the rule does state that additional hemp is necessary to support the growing CBD market, and it notably put pressure on FDA by stating that if “FDA does not provide clarity about their plans for future regulation of CBD, there will continue to be uncertainty and downward pressure on the CBD portion of the hemp market.”
So what does the USDA rule do? Under the USDA rule, states and tribes will have the option of either submitting a proposed hemp regulation plan to the USDA for approval or agreeing to submit to the USDA’s general requirements. All state and tribal plans must include certain provisions, including but not limited to:
Land used for production: State and tribal plans must identify a process for collecting, storing and maintaining relevant information regarding land used for growing hemp in the state. This includes information regarding the description, acreage, and boundaries of the farm land.
Sampling and testing for delta-9 tetrahydrocannabinol (THC): State and tribal plans must implement testing procedures to ensure that plants do not exceed THC levels above 0.3% (as provided in the 2018 Farm Bill). All testing facilities must be DEA approved, as non-compliant product with THC levels over 0.3% would be considered “marihuana” and a schedule 1 substance under the Controlled Substances Act of 1970 (CSA). Additionally, laboratories will be required to report a “measure of uncertainty” in their testing, designed to provide a buffer for the potential variation in sampling and testing procedures. Accordingly, plants testing higher than 0.3% THC but still within the “measure of uncertainty” will be considered compliant.
Disposal of non-compliant products: States and tribes must develop a procedure for destroying non-compliant cannabis containing more than 0.3% THC. Because non-compliant product is considered a controlled substance, all product must be disposed of in a manner consistent with the CSA. Therefore, product must be collected and destroyed by a DEA agent or law enforcement officer.
Inspection of hemp producers: States and tribes must develop procedures for inspecting hemp producers on an annual basis and also for inspecting random samples. The state must also develop procedures to identify and attempt to correct certain negligent acts such as not obtaining licenses or producers exceeding acceptable hemp THC levels.
Information sharing: State and tribal plans must include procedures for reporting information to the USDA. This information must be provided to the USDA within 30 days of receipt from the hemp producers and includes contact information for all hemp producers in the state, legal descriptions of the land used for hemp production, and the license status of all hemp producers in the state.
In states and tribes without an approved or proposed plan, hemp producers will be subject to the USDA general plan. The general plan also provides similar requirements for the testing and sampling of hemp. The USDA will provide licenses directly to hemp producers in states without an approved or submitted plan as some states may not want to have primary regulatory authority of hemp. These states will essentially hand over regulatory responsibility to the USDA. These licenses will be available by application 30 days after the final rule is published. Notably, the draft USDA rule also provides that states and tribes are restricted from prohibiting the transportation or shipment of hemp or hemp products produced under a state plan, tribal plan or a license issued under the FDA. The interstate commerce provision should put an end to the arrests of those transporting legally produced hemp from one state to another. For example, in July 2019, a trucker was arrested and charged with felony possessions of marijuana and intent to distribute while transporting legally grown hemp through South Dakota (South Dakota still considers hemp a controlled substance).
So while the USDA rule is much anticipated and grabbed the attention of many when published, food and dietary supplement manufacturers, distributors, and retailers are still stuck where they were before. We will all continue to wait and see what FDA will do.
During the past year, the headlines have been filled with stories of foodborne illness, product recalls, and consumers becoming sick from tainted food. In a Q&A with Food Safety Tech, Sean O’Leary, CEO at FoodLogiQ, talks food safety, traceability, and how small percentages can translate into big victories for the food industry and for the people they serve.
Food Safety Tech: From your perspective, what is the current sentiment of consumers with regard to food safety?
Sean O’Leary: Over the last few years, the consumer mindset has changed about food in general. We’ve watched fad diets come and go; however, the interest in healthy ingredients and the concern about where food comes from has graduated from a passing trend to a full shift into the public consciousness. Consumers are much more discerning about what they eat; they also demand to know where their food comes from, how it was produced, and how it got to their table. We are living in the age of transparency, and consumer expectations are high.
And who can blame them? CDC statistics tell us that approximately 48 million people get sick every year from foodborne illnesses—and that’s just in the United States; 128,000 of them end up in the hospital. When a person is admitted to the hospital, it affects more than just that one individual. If the patient is the sole breadwinner of their family, their illness affects the entire family. If the person who gets sick is a child, there can be long-term consequences that trickle down to his or her whole community. And when you consider that 3,000 people die every year from foodborne illness—that’s one 9/11 every year. That’s unacceptable, because this is a preventable issue, and unfortunately, these illnesses are an underreported public health problem.
My challenge to the food industry is simple: What if we made just a 1% improvement in the number of cases of foodborne illness? That seems like such a small percentage, but when you do that math, that’s 480,000 people who don’t get sick this year; 1,280 people who aren’t admitted to the hospital; and 30 people who don’t die. Those are significant numbers.
Sean O’Leary joined FoodLogiQ as CEO in January 2019 with more than 25 years of experience in the technology industry.
FST: To help shed additional light on this subject, FoodLogiQ conducted a national survey to tap into how U.S. consumers feel about issues related to food transparency. What did you learn from those consumer responses?
O’Leary: We polled more than 2,000 people to gauge their sentiment around food traceability and their expectations for food companies regarding foodborne illness and product recalls. The survey also posed questions around consumer preferences regarding their food sources and how they are identified on food labels and menus. The results were enlightening, to say the least.
We learned that a brand or restaurant will pay a high price in terms of customer loyalty if they experience a food recall due to consumer illness. And those customers have some strong opinions regarding how quickly the brand or restaurant should address a food safety issue.
35% of survey respondents told us they would avoid an affected brand or restaurant for a few months, and maybe they would return after the issue had been resolved. Meanwhile, nearly 25% admitted they would never use the brand or visit the restaurant again.
Of the respondents who say they care about the quality of the food they eat, 55% say they expect a recall to be executed within 24 to 48 hours.
In reality, it sometimes takes weeks for a product to be pulled from the store or restaurant. This is frequently due to communication issues, since everyone along the supply chain—the grower, supplier, packing and distribution centers, corporate office, and the retailer or restaurant—all must be notified, and a recall plan must be set in motion. Unfortunately, that communication process takes time. When that communication takes place via email or by phone call, the people responsible for pulling product may not have the information they need or may have received misinformation. This can result in lag time, and potentially unsafe product can still get into the hands of consumers.
The faster a food company can address a recall situation and return to business as usual, the faster customers will come back. But comprehensive supply chain transparency is needed to be able to make swift, accurate decisions during this time of crisis. By having a robust end-to-end traceability program and technology that provides real-time data and visibility, companies facing a recall can isolate and surgically withdraw the tainted product out of the supply chain without recalling more items than necessary. That limits the disruption and the waste of good food, which saves the company money.
O’Leary: FoodLogiQ was honored to have the opportunity to share our intricate knowledge of the food supply chain, as well as best practices regarding whole chain traceability during this monumental meeting with the FDA with more than 250 food industry leaders.
In retrospect, one thing is clear—we’re in the midst of a pivotal time of change for the world’s food supply chain. In the United States, the food industry remained status quo for decades, but the introduction of FSMA has brought increased scrutiny and accountability; I think it’s made every food company pause and evaluate where they are with regard to food safety, and that’s a good thing. And now, with the launch of the “New Era” campaign, we’re coming together in a collaborative fashion to map out how technology tools, prevention measures, new business models, and an evolving culture of food safety can be merged as a framework for a long term food safety solution. I agree with the FDA; ‘Smarter Food Safety’ is people-led, FSMA-based, and technology-enabled. It will take all of us working together to reach that goal.
Today The White House issued a statement in which President Trump announced his plan to nominate Stephen Hahn, M.D. to the position of FDA commissioner. Hahn is a radiation and medical oncologist, and currently serves as the chief medical executive at The University of Texas MD Anderson Cancer Center in Houston.
According to The Washington Post, current Acting FDA Commissioner Ned Sharpless will return to his position as director of the National Cancer Institute while HHS official Brett Giroir will step in at FDA until Hahn is confirmed. The Post reports that Sharpless’ term ends Friday, and it could only be extended if the White House named a successor—however, the White House cannot do so because paperwork has not been completed.
Honey is defined as “the natural sweet substance produced by honey bees” from the nectar of plants. However, there is not currently an FDA standard of identity for honey in the United States, which would further define and specify the allowed methods of producing, manufacturing and labeling honey (there is, however, a nonbinding guidance document for honey). Some of the details of honey production that a standard of identity might address include allowable timing and levels of supplemental feeding of bees with sugar syrups and the appropriate use of antibiotics for disease treatment.
In circumstances where strict regulatory standards for foods are not available, they may be created by other organizations.
What Is a Food Standard?
A food standard is “a set of criteria that a food must meet if it is to be suitable for human consumption, such as source, composition, appearance, freshness, permissible additives, and maximum bacterial content.”1
To ensure quality, facilitate trade, and reduce fraud, everyone in the supply chain must have a shared expectation of what each food or ingredient should be. Public standards set those expectations and allow them to be shared. They help ensure that stakeholders have a common definition of quality and purity, as well as the test methods and specifications used to demonstrate that quality and purity. Public standards help ensure fair trade, quality and integrity in food supply chains.
How Is a Standard Different from a Method?
A method is generally an analytical technique to assess a particular property of the content or safety of a food or food ingredient. For example, methods for detection of nitrates in meat products or baby food, coliforms in nut products, or high fructose syrups in honey. Methods are an important component of food standards.
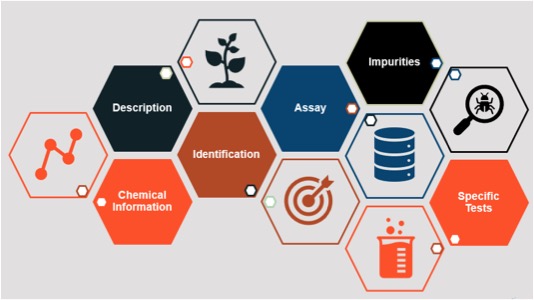
A food standard goes a step further and provides an integrated set of components to define a substance and enable verification of that substance. Standards generally include a description of the substance and its function, one or more identification tests and assays (along with acceptance criteria) to appropriately characterize the substance and ensure its quality, a description of possible impurities and limits for those impurities (if applicable), and other information as needed (see Figure 1).
Figure 1. The Anatomy of an FCC Standard (Source: Food Science Program, Food Chemicals Codex, USP)
Figure 1. The Anatomy of an FCC Standard (Source: Food Science Program, Food Chemicals Codex, USP)
A standard defines both what a food or food ingredient should be and documents how to demonstrate compliance with that definition.
Public Standards and Food Fraud Prevention
Many of the foods prone to fraud are those that are not simple food ingredients, but agricultural products that can be more complex to characterize and identify (such as honey, extra virgin olive oil, spices, etc.). Milk products are an example of a commodity that is prone to fraud with a wide range of adulterants (for example, fluid cow’s milk is associated with 155 adulterants in the Food Fraud Database). Ensuring the quality and purity of a product link milk requires implementation of multiple analytical techniques or the development of non-targeted methods.
The creation of effective public standards with input by a range of stakeholders will be particularly important for ensuring the quality, safety and accurate labeling of these high value commodities in the future.
Reference
A Dictionary of Food and Nutrition 2005, Oxford University Press.
Resources
The Food Chemicals Codex is a source of public standards for foods and food ingredients. It was created by the U.S. FDA and the National Institute of Medicine in 1966 and is currently published by the nonprofit organization USP. The FCC contains 1250 standards for food ingredients, which are developed by expert volunteers and posted for public comment before publication.
The Decernis Food Fraud Database is a continuously updated collection of food fraud records curated specifically to support vulnerability assessments. Information is gathered from global sources and is searchable by ingredient, adulterant, country, and hazard classification. Decernis also partners with standards bodies to provide information about fraudulent adulterants to support standards development.
Food defense is the effort to protect food from intentional acts of adulteration where there is an intent to cause harm. Like counterterrorism laws for many industries, the IA rule, which established a compliance framework for regulated facilities, requires that these facilities prepare a security plan—in this case, a food defense plan—and conduct a vulnerability assessment (VA) to identify significant vulnerabilities that, if exploited, might cause widescale harm to public health, as defined by the FDA. Lessons learned during the conduct of food defense vulnerability and risk assessments and the preparation of the required food defense plan are detailed throughout this three-part series of articles. Part I of this series is intended to assist facilities that have not yet conducted vulnerability assessments or wish to review those already conducted, by leveraging lessons learned from assessments conducted for the largest and most complex global food and beverage facilities.
Lesson 1: VA outcomes are greatly enhanced if a physical security professional is consulted. In support of this contention, there are several physical security mitigation strategies, which can be employed to support a food defense program, that are frequently under-utilized and are not optimally managed by non-security staff. Also, the FDA seems to promote the use of cameras even though this equipment is unlikely to prevent an incident of intentional adulteration. For organizations that choose to use video surveillance, a competent security professional can help organizations engineer and operate video surveillance for maximum benefits and to meet challenging record-keeping requirements when this mitigation strategy is included in a food defense plan.
Lesson 2: Given the focus by the FDA on the insider, a formal insider threat detection program is highly recommended. Trying to promote the common, “See Something, Say Something” strategy may not be enough. For example, if employees are not clearly told what to look for in terms of uniform requirements, how to identify persons who do not belong or changes to a coworker’s baseline behavior, which may indicate moving toward a path to violence or sabotage, then “See Something, Say Something” may end up being no more than a catchy slogan.
A key element of an insider threat detection program is the completion of effective background checks for all persons who will be allowed in the facility unescorted. This includes temporary employees and contractors. A common theme in many of the recent, serious intentional adulteration incidents was that the person responsible was involved in some sort of grievance observable to coworkers and supervisors. In all insider threat detection programs, the grievance becomes an important trip wire. The Carnegie Mellon University Software Engineering Institute has published a document titled, “Common Sense Guide to Mitigating Insider Threats, Sixth Edition”. In this document is some particularly helpful guidance that can be used to stand up an insider threat detection program, but this is an effort that can take some time to fully implement.
Lesson 3: The FDA has made it abundantly clear that they believe the focus for the food and beverage industry should be the radicalized insider. A closer look at all the recently publicized contamination events suggests that there are other profiles that need to be considered. A good foundational model for building profiles of potential offenders can be found in the OSHA definitions for workplace violence offenders, which has been expanded to address ideologically based attacks. Table I applies those descriptions to the food and beverage industry, with an asterisk placed by those offender profiles that exist in recent incidents and discussed later in the text.
Class
OSHA Workplace Violence Offender Description
Motivation Translated to the Food and Beverage Industry
1
The offender has no legitimate relationship to the business or its employee(s). Rather, the violence is incidental to another crime, such as robbery, shoplifting, trespassing or seeking social media fame.
Behavioral Health Patient *
Social Media Fame Seeker *
Copycat *
Extortion *
Economic motivation *
2
The violent person has a legitimate relationship with the business—for example, the person is a customer, client, patient, student, or inmate—and becomes violent while being served by the business, violence falls into this category.
My load isn’t ready, you are costing me money
3
The offender of this type of violence could be a current employee or past employee of the organization who attacks or threatens other employee(s) in the workplace.
I am upset with a coworker and adulterate to create problems for that person *
I am upset with the company and adulterate as retribution and to harm the brand *
Youthful stupidity
I am not paid enough *
4
The offender may or may not have a relationship with the business but has a personal (or perceived personal) relationship with the victim.
I am upset with an intimate partner/ coworker and adulterate to create problems for that person
5
Ideological workplace violence is directed at an organization, its people, and/or property for ideological, religious or political reasons. The violence is perpetrated by extremists and value-driven groups justified by their beliefs.
Radicalized Insider
Table I. A description of OSHA workplace violence offenders and how it can be applied to the F&B industry.
A supermarket in Michigan recalled 1,700 lbs. of ground beef after 111 people fell ill with nicotine poisoning. The offender, an employee, mixed insecticide into the meat to get his supervisor in trouble. In Australia, the entire strawberry industry was brought to its knees after a disgruntled supervisor “spiked” strawberries with needles. There were more than 230 copycat incidents impacting many companies. A contract employee in Japan, apparently disgruntled over his low pay, sprayed pesticide on a frozen food processing line resulting in illnesses to more than 2,000 people. A contract worker upset with a union dispute with the company at a food manufacturing plant videoed himself urinating on the production line, then uploaded the video to the Internet. Be cognizant of any grievances in the workplace and increase monitoring or take other proactive steps to reduce the risk of intentional adulteration.
Lesson 4: The IA Rule requires that every point, step and procedure be analyzed to determine if it is an actionable process step (APS). The Hazard Analysis Critical Control Point flow charts are a good starting point to comply with this element of the law but cannot be counted on completely to achieve the standard of analyzing every point, step or procedure. Critical thinking and persons familiar with the production process need to be involved to ensure that no steps are missed. Oftentimes companies modify the HACCP flow diagrams after a VA.
Lesson 5: The FDA states in the second installment of guidance (here’s the full copy) to the industry that, “There are many possible approaches to conducting a VA. You may choose an approach based on considerations such as the time and resources available and the level of specificity desired. You have the flexibility to choose any VA approach, as long as your VA contains each required component (21 CFR 121.130).”
The FDA further states that the Key Activity Type, or KAT method, is an appropriate method for conducting a VA because it reflects consideration of the three required elements and the inside attacker. Using this methodology alone, however, can result in substantially more APS’s, which might otherwise be ruled out for practical purposes such as a lack of accessibility or a lack of feasibility to contaminate the product at a point, step or procedure. We have experienced up to a 90% decline in APS’s by utilizing another FDA recommended assessment approach, the hybrid approach, which assesses each point, step or procedure as first whether it is a KAT. Then to qualify as an APS, it must also trigger positively for public health impact, accessibility and feasibility to contaminate the product.
Organizations who have yet to execute vulnerability assessments (due July 26, 2020) or who may wish to reflect back on their existing VA’s in an effort to eliminate unnecessary APS’s should find these strategies helpful to focus limited resources to the areas where they can have the greatest effect. The next two articles in this series will cover more information on electronic access, the value of site tours, comparisons to drinking water security strategies, dealing with multi-site assessments and more. Read Part II of this series on intentional adulteration.
Industry from the public and private sector joined for a record turnout during the FDA public meeting yesterday to discuss the agency’s initiative, a new era of smarter food safety. The meeting, which was at maximum capacity for both in-person as well as webcast attendance, began with a call to action from FDA Deputy Commissioner, Office of Food Policy and Response, Frank Yiannas on the importance of all stakeholders in the industry to work together to drive the change. As Yiannas has previously commented, the food industry is in the midst of a revolution. The world is changing faster than ever, and the FDA is challenged with not just creating a safer, more technology-centric and traceable food system, but also getting there faster and more effectively. “I’ve always believed that words we use are important,” he said. As the day’s various discussions would be around the new era of smarter food safety, Yiannas gave the audience a definition to consider: “A new era is a memorable or important date or event, especially one that begins with a new period in our history.”
FDA held breakout sessions centered on areas critical to the initiative:
Tech-enabled traceability and outbreak response
Smarter tools and approaches for prevention
Adapting to new business models and retail modernization
Food safety culture
During each session, FDA facilitators asked the audience questions. The following are some key points brought out during the breakouts.
Tech-Enabled Traceability and Outbreak Response
FDA should consider all parts of the supply chain when thinking about traceability
Take into account considerations for sharing sensitive data along the supply chain
Speaking a common language and creating data standards, along with necessary minimum data elements for traceability is critical
Better communication related to data sharing as well as more meetings with FDA and stakeholders, especially during outbreaks
Show industry the ROI of the data
Provide a roadmap or recommendation for companies on where they can begin on their traceability journey
Request for unity across government agencies (i.e., FDA, USDA), as it would provide more clarity during an outbreak
Smarter Tools and Approaches for Prevention
Trust and transparency are key
Safeguards that address privacy concerns and liability
Data
Data sharing: Concern about retroactive investigations
Types of data: With the “treasure trove” of existing data out there, which is the most important and helpful in improving food safety?
Environmental assessments and root cause analysis—more dialogue between FDA and industry
Adapting to New Business Models and Retail Food Safety Modernization
More need for collaboration
Globalization and use of best practices
Establishing a common standard to level the playing field
Establish best practices for tamper resistance
The last mile: Food safety training for food delivery personnel as well as harmonization for last mile delivery
More consumer education
Food Safety Culture
Emphasis on behavior and humanizing the work: Focusing on what happens within organizations at all levels
Clarity and communication are important
Leveraging current food safety culture best practices as well as any relevant existing standards (i.e., ISO, Codex)
Partnerships are critical, finding the balance between compliance and collaboration
Other Factors FDA Must Consider
The FDA meeting also included panel discussions that drew out the realities FDA must consider in this rapidly changing environment. “These are exciting times and this initiative is recasting our thinking in a whole new light,” said CFSAN Director Susan Mayne, adding, “We need to get ahead of these challenges and not be in reactive mode.”
Consumer awareness and demands for healthy, locally sourced and minimally processed food, for example, are creating increased pressures on food companies and retailers. In addition, the digital savvy and diverse Generation Z (the population born between 1990 and 2010, which will comprise nearly 40% of the U.S. population by 2020) has buying habits and a strong desire for transparency that is shifting how food companies will need to do business, according to Mary Wagner, president of MX Wagner & Associates.
“Trust represents safety, quality and commitment on a much more personal level to our consumers,” said Dirk Herdes, senior vice president at the Nielsen Company, emphasizing the need to communicate with authenticity. “Consumers have never been more informed, but never have been more overwhelmed with information. It’s not data—it’s trust. Trust is the new currency with which we’ll operate.”
FDA and USDA also remain committed to building a stronger relationship between the agencies, said Mindy Brashears, Ph.D., deputy undersecretary for food safety at USDA. “As science moves forward, we have to allow our policies to move forward to keep consumers safe,” she added.
The comments shared during yesterday’s meeting, along with written and electronic comments (with a deadline of November 20), will be considered as FDA puts together its blueprint document for a new era of smarter food safety. More information about providing comments can be found on the Federal Register page.
Last week’s seventh annual Food Safety Consortium brought together a variety of industry experts to discuss key topics around regulation, compliance, leadership, testing, foodborne illness, food defense and more. The following are just a few sound bytes from what we heard at the event. (Click on any photo to enlarge)
“The food system today, while it’s still impressive, it still has one Achilles heel—lack of traceability and transparency.” – Frank Yiannas, deputy commissioner for food policy & response, FDA. Read the full article on Yiannas’ keynote session
“A typical food company only has about 5% visibility into known supply chain threats.” – Ron Stakland, senior business development, FoodChain ID, Inc.
“For most of us, our supply chain is a big black hole. Why are we so fearful of technology? Is it the implementation itself? What if technology could help us solve some of those perennial problems? There are resources available to help us get there.” – ¬ Jeremy Schneider, business development director, food safety and quality assurance, Controlant
“The records tell the story of how well the facility is being managed. It’s the first thing the regulators are going to look at.” – Glenn Black, Ph.D., associate director for research, CFSAN, FDA, on validation considerations and regulations for processing technologies in the food industry
“We’ll see more robotics enter the food space.” – Gina Nicholson Kramer, executive director, Savour Food Safety International
“Changes are happening; you can choose to face it or ignore it. We’re at least 10 years behind on technology. Automation/technology is not a new term in aerospace, etc., but to us [the food industry], it is. We will get there.” – Melody Ge, head of compliance, Corvium, Inc., on how industry should prepare for the data-driven transformation occurring in the smarter era of food safety
It’s okay to risk and fail, but how are going to remediate that with your employee? The more learners practice in different scenarios, the less they rely on specific examples. [They] become more adept with dealing with decision making.” – Kathryn Birmingham, Ph.D., VP for research and development, ImEpik, on employee training
“As a contract lab with the vision of testing for foodborne viruses for about 10 years—it wasn’t until about three or four years ago that we had the test kits to turn that into a reality. We also didn’t have a reference method.” – Erin Crowley, chief scientific officer, Q Laboratories, on the viral landscape of testing in the food industry
“You have to be strong and you have to believe in yourself before you get into any situation—especially as a food safety professional.” – Al Baroudi, Ph.D., vice president of quality assurance and food safety at The Cheesecake Factory, on what it takes to earn respect as a food safety professional
“’See something, say something’ is likely not enough. We recommend that companies develop a formal detection program that includes management buy-in, HR and governance, and policy documents, formal training and an awareness program…While FDA focuses on the insider threat, we feel that using a broader mitigation approach works best.” – R. Spencer Lane, senior security advisor, Business Protection Specialists, Inc. on lessons learned from food defense intentional adulteration vulnerability assessments
“Food safety is a profession, a vocation, [and] a way of life.” – Bob Pudlock, president of Gulf Stream Search
The industry has evolved quite a bit since FSMA was passed eight years ago, and there’s been an overarching recognition that more modern methods to addressing food safety challenges—especially traceability—are essential. “The agency is at the threshold of a sea change in how we’re going to oversee food safety in the nation’s food supply,” said Frank Yiannas, deputy commissioner for food policy & response at FDA at the 2019 Food Safety Consortium. The first driver is the FDA’s new initiative announced earlier this year, coined the “New Era of Smarter Food Safety”. The agency will be looking at how new and emerging technologies can help advance food safety.
“I believe that we’re in the midst of a food revolution,” said Yiannas, pointing to the level of investment in food globally. “Products will be reformulated… new food sources and production approaches will be realized, and the food system will become increasingly digitized.” It will be the job of food safety professionals to adapt to this changing landscape and to ensure that innovation does in fact happen (and safely) in order to feed the growing population.
FDA is taking a new mindset that builds on the success of FSMA but also leverages a people-led, technology enabled method to get there. Just a few days ago the agency launched FDA-TRACK, a new food safety dashboard through which FDA intends to track and measure the performance of the seven FSMA rules—and these measurements will be publicized and available for all stakeholders. Initial available metrics will be tracking outcomes for the FSMA Preventive Controls and the Foreign Supplier Verification Program related to inspections and recalls.
The second driver of this sea change at the FDA involves a shift in its current approach to FSMA and the evolution of the regulation. While the agency continues to educate while it regulates, this past summer FDA took actions that are indicating a shift in its approach to FSMA compliance. On July 30, FDA issued its first warning letter using FSVP authorities to a tahini importer for lack of FSVP compliance. This was followed a month later by actions surrounding the most recent Salmonella outbreak linked to imported papaya. “If you pause and look back, there have been eight outbreaks in eight years,” said Yiannas. The outbreak involved 500 documented cases, 100 hospitalizations and two deaths. “I thought, enough is enough,” he added, and this prompted Acting FDA Commissioner Ned Sharpless, M.D. and Yiannas to issue a statement asking the papaya industry to do more—and urged them to work together to review their practices and make necessary changes to ensure that the papayas they’re offering to the public are safe. The FDA also issued a warning letter to the papaya importer, with the possibility of barring the company for the next five years. “When there’s a public health hazard, FDA will act decisively,” said Yiannas, stating that parity and oversight is important, and domestic and imported food must be safe. “We regulators, we’re going to strike the right balance, and are committed and as passionate as ever in trying to bend the foodborne illness curve.”
Yiannas added that over the next eight years, the trajectory of papaya-related illness will look very different from the last eight years, thanks to the adoption of better technologies—and that’s part of what the FDA’s New Era of Smarter Food Safety is about. “The food system today, while it’s still impressive, it still has one Achilles heel—lack of traceability and transparency,” said Yiannas. There’s a lack of knowledge in where food comes from, where it’s produced, and the fact that many companies simply don’t know as much about their supply chains as they think. With FSMA, Congress anticipated the need to track and trace food, and now the agency is exploring how new technology can enhance tracking and tracing. With regards to tracking foodborne illnesses, the food industry has been in a race between detection and prevention. “We’re getting so good at finding the needle in the haystack, but we can’t find the haystack,” said Yiannas. “We have to provide the same level of investment for technologies for trace backs….we will do that together.”
Frank Yiannas, FDA deputy commissioner for food policy & response, addresses the ways that the public and private sector must work together as part of the agency’s initiative, the New Era of Smarter Food Safety during the 2019 Food Safety Consortium (Image Credit: amybcreative)
When looking at implementing smarter tools and approaches for prevention, the industry needs to work together at not just collecting the data but also converting this data into actionable information. Yiannas emphasized that the agency is not chasing the shiny object—new technology will not be a distraction; it will help solve industry problems and address the new issues that arise with the evolving food system (i.e., cell-cultured meat, plant-based meat, etc.). The agency is also holding a public meeting on October 21 to encourage discussion between the public and private sector, all surrounding this new initiative on smarter food safety.
“What’s become clear to me is [that] there’s so much we an do working together—the public and private sector… Think. Think about how you can do your work differently,” said Yiannas. “We’re all working for the same boss, the consumer. [We have the] same mission and they’re counting on us.”
Attend the Food Defense Plenary Panel Discussion at the 2019 Food Safety Consortium | Tuesday, October 1, 2019Today FDA released an updated version of its Food Defense Plan Builder in efforts to help companies comply with the International Adulteration FSMA rule. Version 2.0 of the tool includes the following sections to help food facility owners and operators in developing a facility-specific food defense plan:
Facility Information
Process/Product Description
Vulnerability Assessment
Mitigation Strategies
Food Defense Monitoring Procedures
Food Defense Corrective Action Procedures
Food Defense Verification Procedures
Supporting Documents
Signature
The tool is for use on a computer, and FDA states that it does not have access to any content or documents used with the tool, nor does it track or monitor how the tool is being used. The agency also emphasizes that use of this tool is not required by law and its use does not mean that a company’s food defense plan is FDA approved or compliant with the IA rule requirements.
The original version of this tool was released in 2013. FDA will be conducting a demonstration of the Food Defense Plan Builder v. 2.0 during a webinar on October 10.
This website uses cookies so that we can provide you with the best user experience possible. Cookie information is stored in your browser and performs functions such as recognising you when you return to our website and helping our team to understand which sections of the website you find most interesting and useful.
Strictly Necessary Cookies
Strictly Necessary Cookies should be enabled at all times so that we can save your preferences for these cookie settings.
We use tracking pixels that set your arrival time at our website, this is used as part of our anti-spam and security measures. Disabling this tracking pixel would disable some of our security measures, and is therefore considered necessary for the safe operation of the website. This tracking pixel is cleared from your system when you delete files in your history.
We also use cookies to store your preferences regarding the setting of 3rd Party Cookies.
If you visit and/or use the FST Training Calendar, cookies are used to store your search terms, and keep track of which records you have seen already. Without these cookies, the Training Calendar would not work.
If you disable this cookie, we will not be able to save your preferences. This means that every time you visit this website you will need to enable or disable cookies again.
Cookie Policy
A browser cookie is a small piece of data that is stored on your device to help websites and mobile apps remember things about you. Other technologies, including Web storage and identifiers associated with your device, may be used for similar purposes. In this policy, we say “cookies” to discuss all of these technologies.
Our Privacy Policy explains how we collect and use information from and about you when you use This website and certain other Innovative Publishing Co LLC services. This policy explains more about how we use cookies and your related choices.
How We Use Cookies
Data generated from cookies and other behavioral tracking technology is not made available to any outside parties, and is only used in the aggregate to make editorial decisions for the websites. Most browsers are initially set up to accept cookies, but you can reset your browser to refuse all cookies or to indicate when a cookie is being sent by visiting this Cookies Policy page. If your cookies are disabled in the browser, neither the tracking cookie nor the preference cookie is set, and you are in effect opted-out.
In other cases, our advertisers request to use third-party tracking to verify our ad delivery, or to remarket their products and/or services to you on other websites. You may opt-out of these tracking pixels by adjusting the Do Not Track settings in your browser, or by visiting the Network Advertising Initiative Opt Out page.
You have control over whether, how, and when cookies and other tracking technologies are installed on your devices. Although each browser is different, most browsers enable their users to access and edit their cookie preferences in their browser settings. The rejection or disabling of some cookies may impact certain features of the site or to cause some of the website’s services not to function properly.
Individuals may opt-out of 3rd Party Cookies used on IPC websites by adjusting your cookie preferences through this Cookie Preferences tool, or by setting web browser settings to refuse cookies and similar tracking mechanisms. Please note that web browsers operate using different identifiers. As such, you must adjust your settings in each web browser and for each computer or device on which you would like to opt-out on. Further, if you simply delete your cookies, you will need to remove cookies from your device after every visit to the websites. You may download a browser plugin that will help you maintain your opt-out choices by visiting www.aboutads.info/pmc. You may block cookies entirely by disabling cookie use in your browser or by setting your browser to ask for your permission before setting a cookie. Blocking cookies entirely may cause some websites to work incorrectly or less effectively.
The use of online tracking mechanisms by third parties is subject to those third parties’ own privacy policies, and not this Policy. If you prefer to prevent third parties from setting and accessing cookies on your computer, you may set your browser to block all cookies. Additionally, you may remove yourself from the targeted advertising of companies within the Network Advertising Initiative by opting out here, or of companies participating in the Digital Advertising Alliance program by opting out here.