

Here is a prediction. Within the next year or years, at some time in your daily work life as a food safety professional you will be called upon to either use genomic tools or to understand and relay information based on genomic tools for making important decisions about food safety and quality. Molecular biologists love to use what often seems like a foreign or secret language. Rest assured dear reader, these are mostly just vernacular and are easily understood once you get comfortable with a bit of the vocabulary. In this the fourth installment of our column we progress to give you another tool for your food genomics tool kit. We have called upon a colleague and sequencing expert, Dr. Sanjay Singh, to be a guest co-author for this topic on sequencing and guide us through the genomics language barrier.
The first report of the annotated (labeled) sequence of the human genome occurred in 2003, 50 years after the discovery of the structure of DNA. In this genome document all the genetic information required to create and sustain a human being was provided. The discovery of the structure of DNA has provided a foundation for a deeper understanding of all life forms, with DNA as a core molecule of genetic information. Of course that includes our food and our tiny friends of the microbial world. Further molecular technological advances in the fields of agriculture, food science, forensics, epidemiology, comparative genomics, medicine, diagnostics and therapeutics are providing stunning examples of the power of genomics in our daily lives. We are only now beginning to harvest the fruits of sequencing and using that knowledge routinely in our respective professions.
In our first column we wrote, “DNA sequencing can be used to determine the names, types, and proportions of microorganisms, the component species in a food sample, and track foodborne diseases agents.” In this month’s column, we present a basic guide to how DNA sequencing chemistry works.

DNA sequencing is the process of determining the precise order of four nucleotide bases, adenine or A, cytosine or C, guanine or G, and thymine or T in a DNA molecule. By knowing the linear sequence of A, C, G, and T in a DNA molecule, the genetic information carried in that particular DNA molecule can be determined.
DNA sequencing happened from the intersections of different fields including biology, chemistry, mathematics, and physics.1,2 The critical breakthrough was provided in 1953 by James Watson, Francis Crick, Maurice Wlkins and Rosalind Franklin when they resolved the now familiar double helix structure of DNA.3 Each helical strand was a polynucleotide, which consists of repeating monomeric units called nucleotides. A nucleotide consists of a sugar (deoxyribose), a phosphate moiety, and one of the four nitrogenous bases—the aforementioned A, C, G, and T. In the double helix, the strands run opposite to each other, commonly referred as anti-parallel. Repeating units of base-pairs (bp), where A always pairs with T and C always pairs with G, are arranged within the double helix so that they are slightly offset from each other like steps in a winding staircase. On a piece of paper, the double helix is often represented by scientists as a flat ladder-like structure, where the base pairs (bp) form the rungs of the ladder while the sugar-phosphate backbone form the antiparallel rails (see Figure 1).
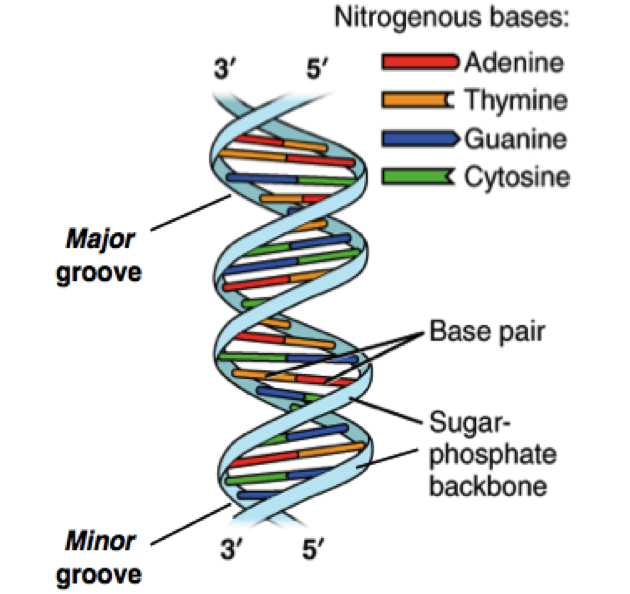
The two ends of each polynucleotide strand are called 5′ or 3′-end, a nomenclature that represents the chemical structure of the deoxyribose sugar at that terminus. The lengths of a single- or double-stranded DNA are often measured in bases (b) or bases pairs (bp), respectively. The two polynucleotide strands can be readily unzipped by heating, and on cooling, the initial double-helix structure is re-formed or re-annealed. The ability to rezip the initial ladder-like structure can be attributed to the phenomenon of base pairing, which merits repetition—the base A always pairs with T and the base G always with C. This rather innocuous phenomenon of base pairing is the basis for the mechanism by which DNA is copied when cells divide and is also the theoretical basis on which most traditional and modern DNA sequencing methodologies have been developed.
Other biological advancements also paved the way towards the development of sequencing technologies. Prominent amongst these were the discovery of enzymes that allowed a scientist to manipulate the DNA. For example, restriction enzymes that recognize and cleave DNA at specific short nucleotide sequences can be used to fragment a long duplex strand of DNA.4 The DNA polymerase enzyme, in the presence of the deoxyribose nucleotide triphosphates (dNTPs: Chemically reactive forms of the nucleotide monomers), can use a single DNA strand to fill in the complementary bases and extend a shorter rail strand (primer extension) of a partial DNA ladder.5 A critical part of the primer extension is the ‘primer’, which are short single-stranded DNA pieces (15 to 30 bases long) that are complementary to a segment of the target DNA. These primers are made using automated high-throughput synthesizer machines. Today, such primers can be rapidly manufactured and delivered on the following day. When the primer and the target DNA are combined through a process called annealing (heat and then cool), they form a structure that shows a ladder-like head and a long single-stranded tail. In 1983, Kary Mullis developed an enzyme-based process called Polymerase Chain Reaction (PCR). Using this protocol, one can pick a single copy of DNA and amplify the same sequence an enormous number of times. One can think of PCR as molecular photocopier in which a single piece of DNA is amplified up to approximately 30 billion copies!
The other critical event that changed the course of DNA sequencing efforts was the publication of the ‘dideoxy chain termination’ method by Dr. Frederick Sanger in December 1977.6 This marked the beginning of the first generation of DNA sequencing techniques. Most next-generation sequencing methods are refinements of the chain termination, or “Sanger method” of sequencing.
Frederick Sanger chemically modified each base so that when it was incorporated into a growing DNA chain, the chain was forcibly terminated. By setting up a primer extension reaction where in one of the chemically modified ‘inactive’ base in smaller quantity is mixed with four active bases, Sanger obtained a series of DNA strands, which when separated based on their size indicated the positions of that particular base in the DNA sequence. By analyzing the results from four such reactions run in parallel, each containing a different ‘inactive’ base, Sanger could piece together the complete sequence of the DNA. Subsequent modifications to the method allowed for the determination of the sequence using dye-labeled termination bases in a single reaction. Since, a sequence of less than <1000 bases can be determined from a single such reaction, the sequence of longer DNA molecules have to be pieced together from many such reads.
Using technologies available in the mid-1990’s, as many as 1 million bases of sequence could be determined per day. However, at this rate, determining the sequence of the 3 billion bp human genome required years of sequencing work. By analogy, this is equivalent to reading the Sunday issue of The New York Times, about 300,000 words, at a pace of 100 words per day. The cost of sequencing the human genome was a whopping $70 million. The human genome project clearly brought forth a need for technologies that could deliver fast, inexpensive and accurate genome sequences. In response, the field initially exploded with modifications to the Sanger method. The impetus for these modifications was provided by advances in enzymology, fluorescent detection dyes and capillary-array electrophoresis. Using the Sanger method of sequencing, one can read up to ~1,000 bp in a single reaction, and either 96 or 384 such reactions (in a 96 or 384 well plate) can be performed in parallel using DNA sequencers. More recently a new wave of technological sequencing advances, termed NGS or next-generation sequencing, have been commercialized. NGS is fast, automated, massively parallel and highly reproducible. NGS platforms can read more than 4 billion DNA strands and generate about a terabyte of sequence data in about six days! The whole 3 billion base pairs of the human genome can be sequenced and annotated in a mere month or less.
Continue to page 2.