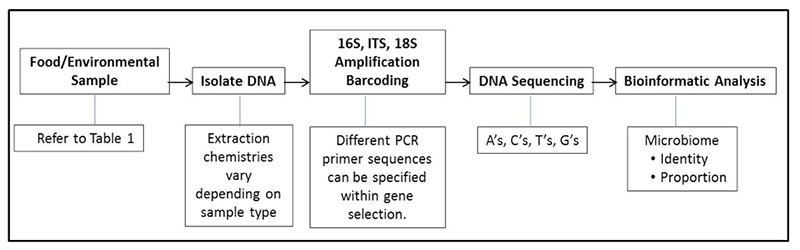
For the past year, Swedish food provider Dafgård has been using a single test to screen each batch of its food for allergens, missing ingredients, and even the unexpected – an unintended ingredient or pathogen. The company extracts DNA from food samples and sends it to a lab for end-to-end sequencing, processing, and analysis. Whether referring to a meatball at a European Ikea or a pre-made pizza at a local grocery store, Dafgård knows exactly what is in its food and can pinpoint potential trouble spots in its supply chains, immediately take steps to remedy issues, and predict future areas of concern.
The power behind the testing is next-generation sequencing (NGS). NGS platforms, like the one my company Clear Labs has developed, consist of the most modern parallel sequencers available in combination with advanced databases and technologies for rapid DNA analysis. These platforms have reduced the cost of DNA sequencing by orders of magnitude, putting the power to sequence genetic material in the hands of scientists and investigators across a range of research disciplines and industries. They have overtaken traditional, first-generation Sanger sequencing in clinical settings over the past several years and are now poised to supplement and likely replace PCR in food safety testing.
For Dafgård, one of the largest food providers in Europe, the switch to NGS has given it the ability to see what was previously impossible with PCR and other technologies. Although Dafgård still uses PCR in select cases, it has run thousands of NGS-based tests over the past year. One of the biggest improvements has been in understanding the supply chain for the spices in its prepared foods. Supply chains for spices can be long and can result in extra or missing ingredients, some of which can affect consumer health. With the NGS platform, Dafgård can pinpoint ingredients down to the original supplier, getting an unparalleled look into its raw ingredients.
Dafgård hopes to soon switch to an entirely NGS-based platform, which will put the company at the forefront of food safety. Embracing this new technology within the broader food industry has been a decade-long process, one that will accelerate in the coming years, with an increased emphasis on food transparency both among consumers and regulators globally.
Transitioning technology
A decade ago, very few people in food safety were talking about NGS technologies. A 2008 paper in Analytical and Bioanalytical Chemistry1 gave an outlook for food safety technology that included nanotechnology, while a 2009 story in Food Safety Magazine2 discussed spectrometric or laser-based diagnostic technologies. Around the same time, Nature magazine named NGS as its “method of the year” for 2007. A decade later, NGS is taking pathogen characterization and food authentication to the next level.
Over the last 30 years, multiple technology transitions have occurred to improve food safety. In the United States, for example, the Hazard Analysis and Critical Control Points (HACCP) came online in the mid-1990s to reduce illness-causing microbial pathogens on raw products. The move came just a few years after a massive outbreak of E. coli in the U.S. Pacific Northwest caused 400 illness and 4 deaths, and it was clear there was a need for change.
Before HACCP, food inspection was largely on the basis of sight, touch, and smell. It was time to take a more science-based approach to meat and poultry safety. This led to the use of PCR, among other technologies, to better measure and address pathogens in the food industry.
HACCP set the stage for modern-era food testing, and since then, efforts have only intensified to combat food-borne pathogens. In 2011, the Food Safety Modernization Act (FSMA) took effect, shifting the focus from responding to pathogens to preventing them. Data from 20153 showed a 30% drop in foodborne-related bacterial and parasitic infections from 2012 to 2014 compared to the same time period in 1996 to 1998.
But despite these vast improvements, work still remains: According to the CDC, foodborne pathogens in the Unites States alone cause 48 million illnesses and 3,000 fatalities every year. And every year, the food safety industry runs hundreds of millions of tests. These tests can mean the difference between potentially crippling business operations and a thriving business that customers trust. Food recalls cost an average of $10M per incident and jeopardize public health. The best way to stay ahead of the regulatory curve and to protect consumers is to take advantage of the new technological tools we now have at our disposal.
Reducing Errors
About 60% of food safety tests currently use rapid methods, while 40% use traditional culturing. Although highly accurate, culturing can take up to five days for results, while PCR and antigen-based tests can be quicker – -one to two days – but have much lower accuracy. So, what about NGS?
NGS platforms have a turnaround of only one day, and can get to a higher level of accuracy and specificity than other sequencing platforms. And unlike some PCR techniques that can only detect up to 5 targets on one sample at a time, the targets for NGS platforms are nearly unlimited, with up to 25 million reads per sample, with 200 or more samples processed at the same time. This results in a major difference in the amount of information yielded.
For PCR, very small segments of DNA are amplified to compare to potential pathogens. But with NGS tools, all the DNA is tested, cutting it into small fragments, with millions of sequences generated – giving many redundant data points for comparing the genome to potential pathogens. This allows for much deeper resolution to determine the exact strain of a pathogen.
Traditional techniques are also rife with false negatives and false positives. In 2015, a study from the American Proficiency Institute4 on about 18,000 testing results from 1999 to 2013 for Salmonella found false negative rates between 2% and 10% and false positive rates between 2% and 6%. Several Food Service Labs claim false positive rates of 5% to 50%.
False positives can create a resource-intensive burden on food companies. Reducing false negatives is important for public health as well as isolating and decontaminating the species within a facility. Research has shown that with robust data analytics and sample preparation, an NGS platform can bring false negative and positive rates down to close to zero for a pathogen test like Salmonella, Listeria, or E.coli.
Expecting the Unexpected
NGS platforms using targeted-amplicon sequencing, also called DNA “barcoding,” represent the next wave of genomic analysis techniques. These barcoding techniques enable companies to match samples against a particular pathogen, allergen, or ingredient. When deeper identification and characterization of a sample is needed, non-targeted whole genome sequencing (WGS) is the best option.
Using NGS for WGS is much more efficient than PCR, for example, at identifying new strains that enter a facility. Many food manufacturing plants have databases, created through WGS, of resident pathogens and standard decontamination steps to handle those resident pathogens. But what happens if something unknown enters the facility?
By looking at all the genomic information in a given sample and comparing it to the resident pathogen database, NGS can rapidly identify strains the facility might not have even known to look for. Indeed, the beauty of these technologies is that you come to expect to find the unexpected.
That may sound overwhelming – like opening Pandora’s box – but I see it as the opposite: NGS offers an unprecedented opportunity to protect against likely threats in food, create the highest quality private databases, and customize internal reporting based on top-of-the-line science and business practices. Knowledge is power, and NGS technologies puts that power directly in food companies’ hands. Brands that adopt NGS platforms can execute on decisions about what to test for more quickly and inexpensively – all the while providing their customers with the safest food possible.
Perhaps the best analogy for this advancement comes from Magnus Dafgård, owner and executive vice president at Gunnar Dafgård AB: “If you have poor eyesight and need glasses, you could be sitting at home surrounded by dirt and not even know it. Then when you get glasses, you will instantly see the dirt. So, do you throw away the glasses or get rid of the dirt?” NGS platforms provide the clarity to see and address problem directly, giving companies like Dafgård confidence that they are using the most modern, sophisticated food safety technologies available.
As NGS platforms continue to mature in the coming months and years, I look forward to participating in the next jump in food safety – ensuring a safe global food system.
Common Acronyms in Food Genomics and Safety
DNA Barcoding: These short, standardized DNA sequences can identify individual organisms, including those previously undescribed. Traditionally, these sequences can come from PCR or Sanger sequencing. With NGS, the barcoding can be developed in parallel and for all gene variants, producing a deeper level of specificity.
ELISA: Enzyme-linked immunosorbent assay. Developed in 1971, ELISA is a rapid substance detection method that can detect a specific protein, like an allergen, in a cell by binding antibody to a specific antigen and creating a color change. It is less effective in food testing for cooked products, in which the protein molecules may be broken down and the allergens thus no longer detectable.
FSMA: Food Safety Modernization Act. Passed in 2011 in the United States, FSMA requires comprehensive, science-based preventive controls across the food supply. Each section of the FSMA consists of specific procedures to prevent consumers from getting sick due to foodborne illness, such as a section to verify safety standards from foreign supply chains.
HACCP: Hazard analysis and critical control points. A food safety management system, HACCP is a preventative approach to quantifying and reducing risk in the food system. It was developed in the 1950s by the Pillsbury Company, the Natick Research Laboratories, and NASA, but did not become as widespread in its use until 1996, when the U.S. FDA passed a new pathogen reduction rule using HACCP across all meat and poultry raw products.
NGS: Next-generation sequencing. NGS is the most modern, parallel, high-throughput DNA sequencing available. It can sequence 200 to 300 samples at a time and generates up to 25 million reads per a single experiment. This level of information can identify pathogens at the strain level and can be used to perform WGS for samples with unknown pathogens or ingredients.
PCR: Polymerase chain reaction. First described in 1985, PCR is a technique to amplify a segment of DNA and generate copies of a DNA sequence. The DNA sequences generated from PCR must be compared to specific, known pathogens. While it can identify pathogens at the species level, PCR cannot provide the strain of a pathogen due to the limited amount of sequencing information generated.
WGS: Whole genome sequencing. WGS uses NGS platforms to look at the entire DNA of an organism. It is non-targeted, which means it is not necessary to know in advance what is being detected. In WGS, the entire genome is cut it into small regions, with adaptors attached to the fragments to sequence each piece in both directions. The generated sequences are then assembled into single long pieces of the whole genome. WGS produces sequences 30 times the size of the genome, providing redundancy that allows for a deeper analysis.
Citations
- Nugen, S. R., & Baeumner, A. J. (2008). Trends and opportunities in food pathogen detection. Analytical and Bioanalytical Chemistry, 391(2), 451-454. doi:10.1007/s00216-008-1886-2
- Philpott, C. (2009, April 01). A Summary Profile of Pathogen Detection Technologies. Retrieved September 08, 2017, from https://www.foodsafetymagazine.com/magazine-archive1/aprilmay-2009/a-summary-profile-of-pathogen-detection-technologies/?EMID
- Ray, L., Barrett, K., Spinelli, A., Huang, J., & Geissler, A. (2009). Foodborne Disease Active Surveillance Network, FoodNet 2015 Surveillance Report (pp. 1-26, Rep.). CDC. Retrieved September 8, 2017, from https://www.cdc.gov/foodnet/pdfs/FoodNet-Annual-Report-2015-508c.pdf.
- Stombler, R. (2014). Salmonella Detection Rates Continue to Fail (Rep.). American Proficiency Institute.