Agriculture and food industries continue to be vulnerable to the complex problems of contamination with natural toxins. Mycotoxins, secondary metabolites produced by fungi, enter the food chain through infection of crops before or after harvest and are typically found in cereals, dried fruits, nuts and spices. Some have well established health impacts, both in humans and animals. A variety of testing solutions exist for mycotoxins, but growth in the use of methods based upon liquid chromatography with tandem mass spectrometry (LC-MS/MS) has enabled the determination of multiple mycotoxins. These methods are extremely sensitive and can be applied to the analysis of raw agricultural commodities, food ingredients and finished products.
Such LC-MS/MS techniques also serve as a powerful tool to investigate the presence of other natural toxins. They have been used for monitoring marine biotoxins[1], and to shed light on the distribution of toxins in terrestrial plants[2], highlighting them as a potentially serious food safety issue. Some terrestrial plants have evolved to produce secondary metabolites as defense mechanisms, which, while beneficial to the plant itself, cause harm to other organisms, including humans. In 2019, a humanitarian food aid product contaminated with tropane alkaloids (TAs) was distributed in Uganda, resulting in a foodborne outbreak which caused over 300 hospitalizations and five deaths[3].
Plant toxins can enter the food chain as constituents of plant products used in food processing or from the seeds and leaves of weeds mixed accidentally with the main food crop at harvest. Low levels of these toxins can be detected in cereals, herbal products, teas, salad crops and some animal products. One important class of plant toxins, pyrrolizidine alkaloids (PAs), are produced by a wide variety of plants commonly belonging to Asteraceae, Fabaceae and Boraginaceae families. Currently, there are more than 660 known PAs and metabolites. They are generally found in products such as honey, pollen, tea, herbal teas, food supplements, spices and aromatic herbs[4]. TAs are another class of plant toxins produced by plants, mostly within the Solanaceae family, and have been found in a range of agricultural cereal crops (e.g. linseed, soybean, millet, sunflower and buckwheat), tea, and herbal blends and infusions[5].
EU Legislation on Plant Toxins
The European Food Safety Authority (EFSA) has published the results of various risk assessments on those plant toxins considered to be the greatest risk to human health[6],[7], leading to the introduction of legislation on plant toxins in food by the European Commission[8]. Maximum levels have been set for PAs in herbs, spices, teas, herbal infusions and pollen products. These, which refer to the sum of 35 specified PAs (including their N-oxidized forms), vary between commodities. For example, the maximum level for PAs in most teas is 150 µg/kg, whereas the value for cumin is set at 400 µg/kg. Although there are more than 200 different TAs known, maximum levels have only been set for atropine and scopolamine (from 0.2 to 50 µg/kg, depending on the commodity). These regulations require that these plant toxins be monitored in specified foods by the Member State Food Safety Authorities and by food business operators, including those imported into the EU.
Access to data from retail surveys for PAs and TAs remains scarce when compared to that which is available for mycotoxins. However, in recent years, the number of food alerts reported on the Rapid Alert System for Food and Feed (RASFF) portal on the occurrence of PAs and TAs in different food products, exceeding maximum levels, has notably increased. The RASFF system was established to ensure the exchange of information between EU member countries to support swift reaction by food safety authorities in case of risks to public health resulting from issues with the food chain. Casado reported levels of PAs related to RASFF alerts with values ranging from 26 to 556,910 µg/kg[9], whereas the highest values of atropine and scopolamine were reported by Goncalves in tea and herbs (mean 173 and 147 µg/kg, respectively)[10]. In relation to consignments of cumin from Türkiye, a high rate of noncompliance with the relevant requirements provided for in EU legislation with respect to contamination by PAs was detected during official controls performed by the Member States[11]. The frequency of mandatory checks to be performed at border control has recently been increased to 30 %[12]. This has prompted greater awareness of the issue in other countries importing into the EU.
Techniques for Measuring Plant Toxins
Sampling plays a crucial part in precise determination of plant toxins levels in a certain lot, as contaminants within a lot may be heterogeneously distributed. It is also necessary to establish general method of analysis performance criteria to ensure that control laboratories use methods of analysis with comparable levels of performance. In December 2023, the European Commission published legislation establishing methods of sampling and analysis for the control of plant toxins levels in food[13].
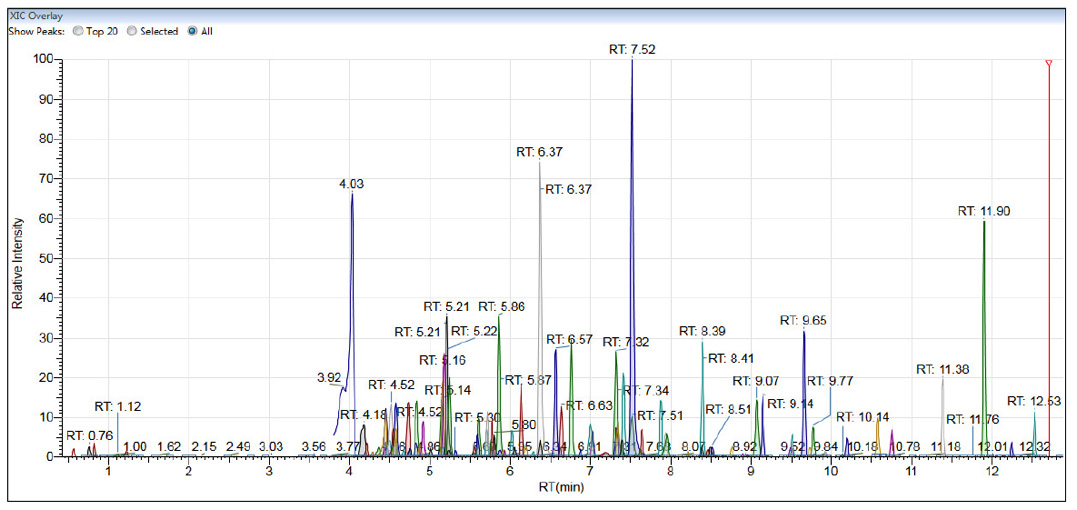
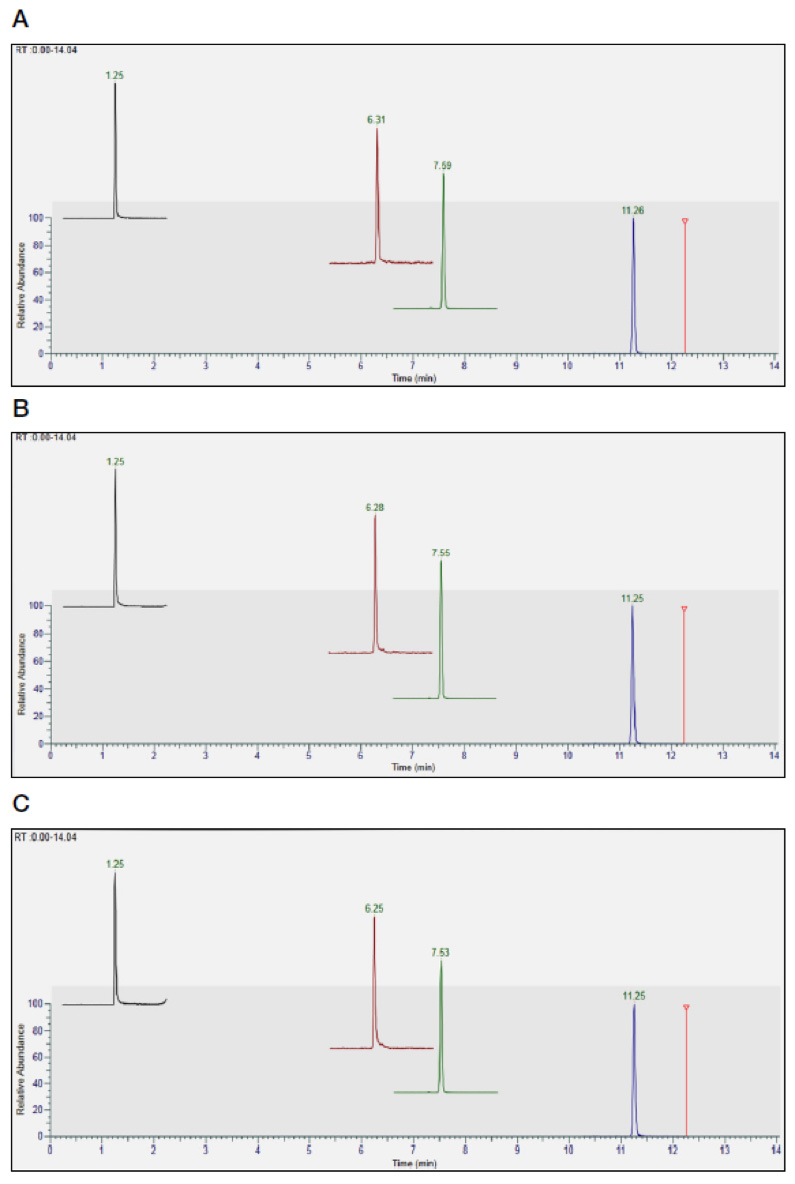
Methods for PAs rely on extraction with acidified water, followed by solid-phase extraction (SPE) using a mixed-mode sorbent, which provides dual retention modes of reversed-phase and cation-exchange, followed by LC-MS/MS using alkaline or acidic chromatographic conditions[14]. The main analytical challenge is the presence of many isomers that are extremely difficult to resolve in the chromatographic dimension and exhibit the same MRM transitions. When attempting analysis in a single chromatographic run, one is left with a few pairs of coeluting isomers, which can be quantified as a sum. TAs are typically extracted with an acidified mixture of water and methanol/acetonitrile (including QuEChERS), followed by LC-MS/MS. Passing the extract through a simple ultrafiltration device or SPE cartridge can remove matrix co-extractives, enhancing method performance. To rationalize analyses in high-throughput laboratory environments, the scope of multi-mycotoxin methods can easily be extended to include the two regulated TAs, atropine and scopolamine[15].
While efforts have been made to address the food safety issue of plant toxins in Europe and reduce risk to the consumer, the number of food alerts seems to be on the rise. Fortunately, challenges with the determination of plant toxins in foods have largely been overcome, enabling testing to be carried out for checking regulatory compliance and monitoring occurrence, ensuring the safety of products for human consumption.
![]()
![]()
References:
[1] Panda D. et al. (2022). Recent advancements in LC-MS based analysis of biotoxins: Present and future challenges. Mass Spec Rev. 41:766-803.
[2] Urugo, M. et al. (2023). Naturally Occurring Plant Food Toxicants and the Role of Food Processing Methods in Their Detoxification. Int. J. Food Sci. 2023 Article ID 9947841, 16pp.
[3] Abia W. et al. (2021). Tropane alkaloid contamination of agricultural commodities and food products in relation to consumer health: Learnings from the 2019 Uganda food aid outbreak. Compr. Rev. Food Sci. Food Saf. 20(1):501-525.
[4] Fuente-Ballesteros A. et al. (2024). Comprehensive overview of the analytical methods for determining
pyrrolizidine alkaloids and their derived oxides in foods. J. Food Compos. Anal. 125:105758.
[5] De Nijs, M. et al. (2023). Emerging Issues on Tropane Alkaloid Contamination of Food in Europe. Toxins 15(2):98.
[6] EFSA (2013). Scientific Opinion on tropane alkaloids in food and feed. EFSA Panel on Contaminants in the Food Chain. EFSA J. 11:3386.
[7] EFSA (2017). Risks for human health related to the presence of pyrrolizidine alkaloids in honey, tea, herbal infusions and food supplements. EFSA J. 15:4908.
[8] European Commission (2023). Commission Regulation (EU) 2023/915 of 25 April 2023 on maximum levels for certain contaminants in food and repealing Regulation (EC) No 1881/2006. OJ L 119:103–157.
[9] Casado, N. et al. (2022). The concerning food safety issue of pyrrolizidine alkaloids: An overview. Trends Food Sci. Technol. 120:123-139
[10] Gonzalez-Gómez L. et al. (2022). Occurrence and Chemistry of Tropane Alkaloids in Foods, with a Focus on Sample Analysis Methods: A Review on Recent Trends and Technological Advances. Foods 11:407.
[11] https://webgate.ec.europa.eu/rasff-window/screen/notification/651495
[12] European Commission (2024). Commission Implementing Regulation (EU) 2024/286 of 16 January 2024 amending Implementing Regulation (EU) 2019/1793 on the temporary increase of official controls and emergency measures governing the entry into the Union of certain goods from certain third countries. OJ L 2024/286.
[13] European Commission (2023). Commission Implementing Regulation (EU) 2023/2783 of 14 December 2023 laying down the methods of sampling and analysis for the control of the levels of plant toxins in food and repealing Regulation (EU) 2015/705. OJ L 2023/2783.
[14] Method Development and Validation for the Determination of Pyrrolizidine Alkaloids in a Range of Plant-Based Foods and Honey Using LC-MS/MS. Waters Application Note 720007624.
[15] Development of a Multi-Toxin UPLC-MS/MS Method for 50 Mycotoxins and Tropane Alkaloids in Cereal Commodities. Waters Application Note 720007476.