FDA has released a report on the multiagency investigation of a Salmonella Typhimurium outbreak associated with packaged salad greens grown in a controlled environment agriculture (CEA) operation. The outbreak, which occurred between June and August 2021, resulted in 31 reported illnesses and four hospitalizations. It is also believed to be the first of its kind associated with leafy greens grown in a CEA facility.
No “conclusive” root cause was found, but the FDA did pinpoint the outbreak strain of Salmonella to a stormwater retention basin located next to the CEA farm. The investigation did not, however, find that this was the definitive source of contamination of the leafy greens. The agency also identified certain conditions, factors and practices that could lead to contamination, including the pond water used, growth media storage methods, water management practices and overall sanitation practices.
In the report, the FDA listed eight requirements and recommendations that apply to hydroponic facilities using CEA, including implementing effective sanitation procedures and sampling plans, conducting pre- and post-harvest sampling and testing of food, water and the physical environment, implementing procedures that are effective in rapidly cooling and cold-holding harvested leafy greens after harvest, and ensuring all growing pond water is safe and of sanitary quality.
Dole Fresh Vegetables, Inc. issued a voluntary recall of certain cases of its garden salad over concern of possible Listeria monocytogenes contamination. Although no illnesses have been reported, the company is pulling select lots of its garden salads marketed under the Dole, Marketside, Kroger and Salad Classics names.
The recall was taken as a precaution after a single sample of garden salad tested positive for Listeria monocytogenes in random sampling conducted by the Department of Agriculture in Georgia.
The company announcement states that the product is beyond its “best if used by” date and should no longer be on store shelves. The products were distributed in Alabama, Florida, Georgia, Louisiana, Massachusetts, Maryland, North Carolina, Pennsylvania, South Carolina and Virginia.
Today Beech-Nut Nutrition Company announced a voluntary recall of one lot of its Stage 1 Single Grain Rice Cereal following sampling that revealed the product tested above the guidance level for naturally occurring inorganic arsenic set by FDA last summer. The routine sampling was conducted by the State of Alaska. The recalled item has an expiration date of May 1, 2022.
“The safety of infants and children is Beech-Nut’s top priority. We are issuing this voluntary recall, because we learned through routine sampling by the State of Alaska that a limited quantity of Beech-Nut Single Grain Rice Cereal products had levels of naturally-occurring inorganic arsenic above the FDA guidance level, even though the rice flour used to produce these products tested below the FDA guidance level for inorganic arsenic,” said Jason Jacobs, Vice President, Food Safety and Quality, Beech-Nut, in a company announcement published on FDA’s website.
Perhaps even bigger news is Beech-Nut’s announcement that it is exiting the market for its branded Single Grain Rice Cereal. The company is concerned that it will not be able to consistently obtain rice flour that is well-below FDA’s guidance level (as well as Beech-Nut’s specifications) for naturally occurring inorganic arsenic.
Watch the on-demand complimentary virtual event in our Food Safety Hazards Series, “Salmonella Detection, Mitigation, Control and Regulation” (Original air date: Thursday, July 15, 2021)As part of ongoing surveillance efforts resulting from recurring outbreaks, the FDA announced that it will conduct direct sampling and testing of lettuce grown in the Salinas Valley region of California. From May through November 2021, the agency will test samples for Shiga toxin-producing Escherichia coli (STEC), including E. coli O157:H7, and Salmonella spp. Direct sampling will be conducted at commercial cooling and cold storage facilities where field heat is removed from harvested lettuce and product is cold-stored prior to processing. “Sample collection at commercial coolers helps the FDA efficiently obtain samples from multiple farms at centralized locations and facilitates prompt traceback and follow-up if contamination is detected,” according to a CFSAN update.
FDA laboratories plan to test about 500 post-harvest samples of iceberg, leaf and romaine lettuce, with each sample consisting of 10 subsamples (one head of lettuce that is trimmed, cored or wrapped; or romaine lettuce leaves or one package of hearts).
In compliance with COVID-19 safety practices, the agency investigators will preannounce their visits.
Find records of fraud such as those discussed in this column and more in the Food Fraud Database. Image credit: Susanne Kuehne
Since only 417 Masters of Wine exist globally (and their palates and noses)—and they are amazing in identifying wines by grape varietal or blend, type, vintage and location—it is a good idea to have some automated backup when it comes to wine fraud detection. Aside from other analytical methods, nuclear magnetic resonance (NMR) spectroscopy can be used in the authentication of wine. The new proton measurement 1H NMR Method with easier sample preparation is recommended for the investigation of wine fraud, to detect for example the addition of water or sugar. NMR spectroscopy measures several compounds of a wine at once and therefore is able to detect a fingerprint of a wine, such as the geographic origin or grape varietal.
–UPDATE March 9, 2021 — Today the FDA confirmed that the recalled cheeses were also distributed to Rhode Island. “States with confirmed distribution now include: AL, CT, FL, GA, IA, IL, IN, KS, KY, MA, MD, MI, MN, MO, MS, NC, NJ, NY, NE, OH, PA, RI, SC, TN, VA, and WI.”
–UPDATE February 24, 2021 — FDA has expanded its warning related to El Abuelito Cheese to include all cheese branded by the company “until more information is known”.
—END UPDATE—
A multistate outbreak of Listeria monocytogenes has been linked to Hispanic-style fresh and soft cheeses produced by El Abuelito Cheese, Inc. As a result, the company has recalled all Questo Fresco products with sell by dates through March 28 (032821).
Join Food Safety Tech on April 15 for the complimentary Food Safety Hazards Series: Listeria Detection, Mitigation, Control & Regulation“As the FDA stated, about this outbreak investigation, the Connecticut Department of Public Health collected product samples of El Abuelito-brand Hispanic-style fresh and soft cheeses from a store where a sick person bought cheeses. Sample analysis showed the presence of Listeria monocytogenes in samples of El Abuelito Queso Fresco sold in 10 oz packages, marked as Lot A027 with an expiration date of 02/26/2021,” the company stated in an announcement posted on FDA’s website. “Samples are currently undergoing Whole Genome Sequencing (WGS) analysis to determine if the Listeria monocytogenes found in these samples is a match to the outbreak strain. At this time, there is not enough evidence to determine if this outbreak is linked to El Abuelito Queso Fresco.”.
The recalled products were distributed to Connecticut, Maryland, New Jersey, North Carolina, New York, Pennsylvania and Virginia. Thus far seven people, all of whom have been hospitalized, have fallen ill.
FDA recommends that consumers, restaurants and retailers do not consume, sell or serve any of the recalled cheeses. The agency also states that anyone who purchased of received the recalled products use “extra vigilance in cleaning and sanitizing any surfaces and containers that may have come in contact with these products to reduce the risk of cross-contamination.”
Following a report released nearly two weeks ago about the potential danger posed by toxic heavy metals found in baby foods manufactured by several major companies, FDA has issued a response. The report, “Baby Foods Are Tainted with Dangerous Levels of Arsenic, Lead, Cadmium, and Mercury”, was released by the U.S. House of Representatives Committee on Oversight and Reform Subcommittee on Economic and Consumer Policy on February 4. The Subcommittee stated that FDA should require baby food manufacturers to test their finished products for toxic heavy metals and require any toxic heavy metals be reported on food labeling. It also stated that FDA should set maximum levels of toxic heavy metals allowed in baby foods.
“The FDA has been actively working on this issue using a risk-based approach to prioritize and target the agency’s efforts. Consumers should know that FDA scientists routinely monitor levels of toxic elements in baby foods, along with other foods consumed in the country’s diet, through the Total Diet Study,” the agency stated in a CFSAN update. “Further, the FDA also monitors baby food under the FDA’s compliance program for Toxic Elements in Food and Foodware, and Radionuclides in Food and through targeted sampling assignments.”
FDA cited its work in sampling infant rice cereal for arsenic, which it says has resulted in safer products on the market, along with its recent court order to stop a U.S. company from distributing adulterated juice that had potentially harmful levels of inorganic arsenic and patulin (a mycotoxin).
The CFSAN update, however, did not specifically address the companies or baby foods called out in the Subcommittee’s report.
Today the FDA announced a new plan to collect samples of romaine lettuce as part of its ongoing surveillance after the spring 2018 multistate outbreak of E. coli O157:H7. The samples, which will be tested for Shiga toxin-producing Escherichia coli (STEC) and Salmonella, will be collected from commercial coolers in Yuma County, Arizona during the current harvest season.
FDA plans to collect and test about 500 samples (each of which will consist of 10 subsamples), beginning in February and continuing through the end of the harvest season. In order to reduce the time between sample collection and reporting results, an independent lab close to the collection sites in Arizona will be testing the samples. FDA expects to receive test results within 24 hours.
“Helping to ensure the safety of leafy greens continues to be a priority of the FDA. This assignment adds to other work underway in collaboration with stakeholders in the Yuma agricultural region to implement actions identified in the Leafy Greens Action Plan, including a multi-year study to assess the environmental factors that impact the presence of foodborne pathogens in this region. Consistent with the action plan, the agency will engage with industry on conducting root cause analyses for any positive samples found during this assignment. Root cause analyses are important in that they seek to identify potential sources and routes of contamination, inform what preventive measures are needed, and help prevent outbreaks of foodborne illness,” FDA stated in a release.
COVID-19 precautions will be taken during the sampling plan. Agency investigators will preannounce visits and wear PPE while conducting the work.
Tanimura & Antle issued a voluntary recall of single-head packaged romaine lettuce.
Tanimura & Antle, Inc. is voluntarily recalling its packaged single head romaine lettuce, out of an abundance of caution, due to possible E. Coli 0157:H7 contamination. The product has a packaged date of 10/15/2020 or 10/16/2020, and the UPC number 0-27918-20314-9.
Although no illnesses have been reported, the recall is based on the test result of a random sample taken and analyzed by the Michigan Department of Agriculture and Rural Development. The company distributed 3,396 cartons to 20 states. Retailers and distributors can identify the affected products using the Product Traceability Initiative stickers (571280289SRS1 and 571280290SRS1) that are attached to the exterior of the case.
With the increasing globalization of the food industry, ensuring that products reaching consumers are safe has never been more important. Local, state and federal regulatory agencies are increasing their emphasis on the need for food and beverage laboratories to be accredited to ISO/IEC 17025 compliance. This complicated process can be simplified and streamlined through the adoption of LIMS, making accreditation an achievable goal for all food and beverage laboratories.
With a global marketplace and complex supply chain, the food industry continues to face increasing risks for both unintentional and intentional food contamination or adulteration.1 To mitigate the risk of contaminated products reaching consumers, the International Organization for Standardization (ISO), using a consensus-based approval process, developed the first global laboratory standard in 1999 (ISO/IEC 17025:1999). Since publication, the standard has been updated twice, once in 2005 and most recently in 2017, and provides general requirements for the competence of testing and calibration laboratories.2
In the recent revision, four key updates were identified:
A revision to the scope to include testing, calibration and sampling associated with subsequent calibration and testing performed by a laboratory.3
An emphasis on the results of a process instead of focusing on prescriptive procedures and policies.4
The introduction of the concept of a risk-based approach used in production quality management systems.2
A stronger focus on information technologies/management systems, specifically Laboratory Information Management System (LIMS).4
As modern-day laboratories reduce their reliance on hard copy documents and transition to electronic records, additional emphasis and guidance for ISO 17025 accreditation in food testing labs using LIMS was greatly needed. Food testing laboratories have increased reliance on LIMS to successfully meet the requirements of accreditation. Food and beverage LIMS has evolved to increase a laboratory’s ability to meet all aspects of ISO 17025.
Figure 1. A schematic representation of some of the requirements of ISO/IEC 17025:2017 compliance. (Figure courtesy CloudLIMS)
Traceability
Chain of Custody
A key element for ISO 17025 accredited laboratories is the traceability of samples from accession to disposal.5 Sometimes referred to as chain of custody, properly documented traceability allows a laboratory to tell the story of each sample from the time it arrives until the time it is disposed of.
LIMS software allows for seamless tracking of samples by employing unique sample accession numbers through barcoding processes. At each step of sample analysis, a laboratory technician updates data in a LIMS by scanning the sample barcode, establishing time and date signatures for the analysis. During an ISO 17025 audit, this information can be quickly obtained for review by the auditor.
Procurement and Laboratory Supplies
ISO 17025 requires the traceability of all supplies or inventory items from purchase to usage.6 This includes using approved vendors, documentation of receipt, traceability of supply usage to an associated sample, and for certain products, preparation of supply to working conditions within the laboratory. Supply traceability impacts multiple departments and coordinating this process can be overwhelming. A LIMS for food testing labs helps manage laboratory inventory, track usage of inventory items, and automatically alerts laboratory managers to restock inventory once the quantity falls below a threshold level.
A food LIMS can ensure that materials are ordered from approved vendors only, flagging items purchased outside this group. As supplies are inventoried into LIMS, the barcoding process can ensure accurate storage. A LIMS can track the supply through its usage and associate it with specific analytical tests for which inventory items are utilized. As products begin to expire, a LIMS can notify technicians to discard the obsolete products.
One unique advantage of a fully integrated LIMS software is the preparation and traceability of working laboratory standards. A software solution for food labs can assist a technician in preparing standards by determining the concentration of solvents needed based on the input weight from a balance. Once prepared, LIMS prints out a label with barcodes and begins the supply traceability process as previously discussed.
Quality Assurance of Test and Calibration Data
This section of ISO 17025 pertains to the validity of a laboratory’s quality system including demonstrating that appropriate tests were performed, testing was conducted on properly maintained and calibrated equipment by qualified personnel, and with appropriate quality control checks.
Laboratory Personnel Competency
Laboratory personnel are assigned to a specific scope of work based upon qualifications (education, training and experience) and with clearly defined duties.7 This process adds another layer to the validity of data generated during analysis by ensuring only appropriate personnel are performing the testing. However, training within a laboratory can be one of the most difficult components of the accreditation process to capture due to the rapid nature in which laboratories operate.
With a food LIMS, management can ensure employees meet requirements (qualifications, competency) as specified in job descriptions, have up-to-date training records (both onboarding and ongoing), and verify that only qualified, trained individuals are performing certain tests.
Calibration and Maintenance of Equipment
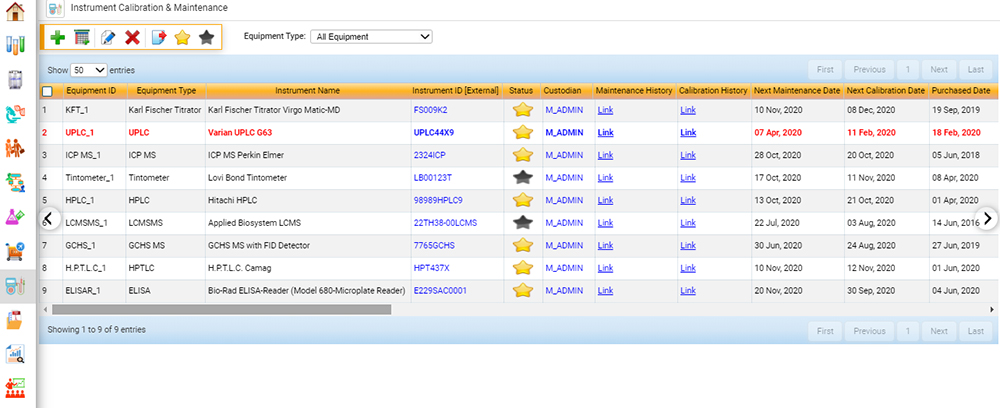
Within the scope of ISO 17025, food testing laboratories must ensure that data obtained from analytical instruments is reliable and valid.5 Facilities must maintain that instruments are in correct operating condition and that calibration data (whether performed daily, weekly, or monthly) is valid. As with laboratory personnel requirements, this element to the standard adds an additional layer of credibility that sample data is precise, accurate, and valid.
A fully integrated software solution for food labs sends a notification when instrument calibration is out of specification or expired and can keep track of both routine internal and external maintenance on instruments, ensuring that instruments are calibrated and maintained regularly. Auditors often ask for instrument maintenance and calibration records upon the initiation of an audit, and LIMS can swiftly provide this information with minimal effort.
Figure 2. A preconfigured food LIMS to manage instrument calibration and maintenance data. (Figure courtesy of CloudLIMS)
Measurement of Uncertainty (UM)
Accredited food testing laboratories must measure and report the uncertainty associated with each test result.8 This is accomplished by using certified reference materials (CRM), or known spiked blanks. UM data is trended using control charts, which can be prepared using labor-intensive manual input or performed automatically using LIMS software. A fully integrated food LIMS can populate control data from the instrument into the control chart and determine if sample data analyzed in that batch can be approved for release.
Valid Test Methods and Results
Accurate test and calibration results can only be obtained with methods that are validated for the intended use.5 Accredited food laboratories should use test methods that are current and contain embedded quality control standards.
A LIMS for food testing labs can ensure correct method selection by technicians by comparing data from the sample accession input with the test method selected for analysis. Specific product identifiers can indicate if methods have been validated. As testing is performed, a LIMS can track time signatures to ensure protocols are properly performed. At the end of the analysis, results of the quality control samples are linked to the test samples to ensure only valid results are available for clients. Instilling checks at each step of the process allows a LIMS to auto-generate Certificates of Analysis (CoA) knowing that the testing was performed accurately.
Data Integrity
The foundation of a laboratory’s reputation is based on its ability to provide reliable and accurate data. ISO 17025:2017 includes specific references to data protection and integrity.10 Laboratories often claim within their quality manuals that they ensure the integrity of their data but provide limited details on how it is accomplished making this a high priority review for auditors. Data integrity is easily captured in laboratories that have fully integrated their instrumentation into LIMS software. Through the integration process, data is automatically populated from analytical instruments into a LIMS. This eliminates unintentional transcription errors or potential intentional data manipulation. A LIMS for food testing labs restricts access to changing or modifying data, allowing only those with high-level access this ability. To control data manipulation even further, changes to data auto-populated in LIMS by integrated instrumentation are tracked with date, time, and user signatures. This allows an auditor to review any changes made to data within LIMS and determine if appropriate documentation was included on why the change was made.
Sampling
ISO 17025:2017 requires all food testing laboratories to have a documented sampling plan for the preparation of test portions prior to analysis. Within the plan, the laboratory must determine if factors are addressed that will ensure the validity of the testing, ensure that the sampling plan is available to the laboratory (or the site where sampling is performed), and identify any preparation or pre-treatment of samples prior to analysis. This can include storage, homogenization (grinding/blending) or chemical treatments.9
As sample information is entered into LIMS, the software can specify the correct sampling method to be performed, indicate appropriate sample storage conditions, restrict the testing to approved personnel and provide electronic signatures for each step.
Monitoring and Maintenance of the Quality System
Organization within a laboratory’s quality system is a key indicator to assessors during the audit process that the facility is prepared to handle the rigors that come with accreditation.10 Assessors are keenly aware of the benefits that a food LIMS provides to operators as a single, well-organized source for quality and technical documents.
Document Control
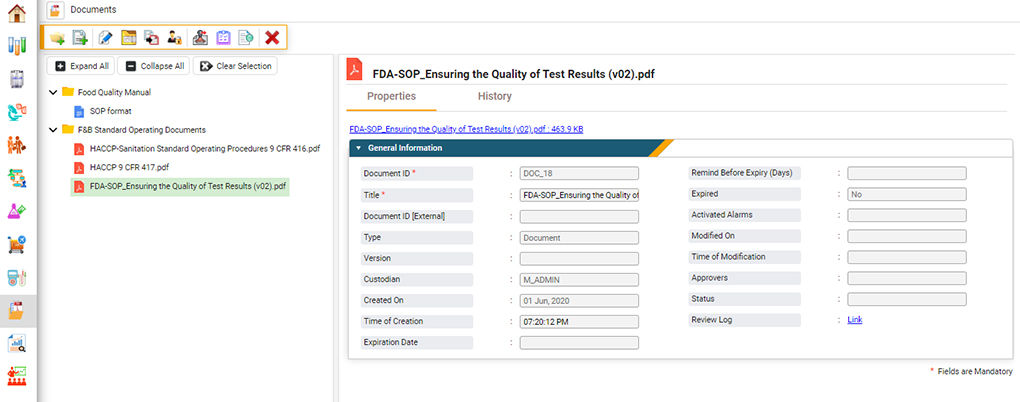
An ISO 17025 accredited laboratory must demonstrate document control throughout its facility.6 Only approved documents are available for use in the testing facility, and the access to these documents is restricted through quality control. This reduces the risk of document access or modification by unauthorized personnel.
LIMS software efficiently facilitates this process in several ways. A food LIMS can restrict access to controlled documents (both electronic and paper) and require electronic signatures each time approved personnel access, modify or print them. This digital signature provides a chain of custody to the document, ensuring that only approved controlled documents are used during analyses and that these documents are not modified.
Figure 3. A software solution for food labs helps manage documents, track their revision history, and ensure document control. (Figure courtesy of CloudLIMS)
Corrective Actions/Non-Conforming Work
A fundamental requirement for quality systems is the documentation of non-conforming work, and subsequent corrective action plans established to reduce their future occurrence.5
A software solution for food labs can automatically maintain electronic records of deviations in testing, flagging them for review by quality departments or management. After a corrective action plan has been established, LIMS software can monitor the effectiveness of the corrective action by identifying similar non-conforming work items.
Conclusion
Food and beverage testing laboratories are increasingly becoming accredited to ISO 17025. With recent changes to ISO 17025, the importance of LIMS for the food and beverage industry has only amplified. A software solution for food labs can integrate all parts of the accreditation process from personnel qualification, equipment calibration and maintenance, to testing and methodologies.11 Fully automated LIMS increases laboratory efficiency, productivity, and is an indispensable tool for achieving and maintaining ISO 17025 accreditation.
Perry Johnson Laboratory Accreditation (2019). An Overview of Changes Between 17025:2005 and 17025:2017. ISO/IEC 17025:2017 Transition. https://www.pjlabs.com/downloads/17025-Transition-Book.pdf
This website uses cookies so that we can provide you with the best user experience possible. Cookie information is stored in your browser and performs functions such as recognising you when you return to our website and helping our team to understand which sections of the website you find most interesting and useful.
Strictly Necessary Cookies
Strictly Necessary Cookies should be enabled at all times so that we can save your preferences for these cookie settings.
We use tracking pixels that set your arrival time at our website, this is used as part of our anti-spam and security measures. Disabling this tracking pixel would disable some of our security measures, and is therefore considered necessary for the safe operation of the website. This tracking pixel is cleared from your system when you delete files in your history.
We also use cookies to store your preferences regarding the setting of 3rd Party Cookies.
If you visit and/or use the FST Training Calendar, cookies are used to store your search terms, and keep track of which records you have seen already. Without these cookies, the Training Calendar would not work.
If you disable this cookie, we will not be able to save your preferences. This means that every time you visit this website you will need to enable or disable cookies again.
Cookie Policy
A browser cookie is a small piece of data that is stored on your device to help websites and mobile apps remember things about you. Other technologies, including Web storage and identifiers associated with your device, may be used for similar purposes. In this policy, we say “cookies” to discuss all of these technologies.
Our Privacy Policy explains how we collect and use information from and about you when you use This website and certain other Innovative Publishing Co LLC services. This policy explains more about how we use cookies and your related choices.
How We Use Cookies
Data generated from cookies and other behavioral tracking technology is not made available to any outside parties, and is only used in the aggregate to make editorial decisions for the websites. Most browsers are initially set up to accept cookies, but you can reset your browser to refuse all cookies or to indicate when a cookie is being sent by visiting this Cookies Policy page. If your cookies are disabled in the browser, neither the tracking cookie nor the preference cookie is set, and you are in effect opted-out.
In other cases, our advertisers request to use third-party tracking to verify our ad delivery, or to remarket their products and/or services to you on other websites. You may opt-out of these tracking pixels by adjusting the Do Not Track settings in your browser, or by visiting the Network Advertising Initiative Opt Out page.
You have control over whether, how, and when cookies and other tracking technologies are installed on your devices. Although each browser is different, most browsers enable their users to access and edit their cookie preferences in their browser settings. The rejection or disabling of some cookies may impact certain features of the site or to cause some of the website’s services not to function properly.
Individuals may opt-out of 3rd Party Cookies used on IPC websites by adjusting your cookie preferences through this Cookie Preferences tool, or by setting web browser settings to refuse cookies and similar tracking mechanisms. Please note that web browsers operate using different identifiers. As such, you must adjust your settings in each web browser and for each computer or device on which you would like to opt-out on. Further, if you simply delete your cookies, you will need to remove cookies from your device after every visit to the websites. You may download a browser plugin that will help you maintain your opt-out choices by visiting www.aboutads.info/pmc. You may block cookies entirely by disabling cookie use in your browser or by setting your browser to ask for your permission before setting a cookie. Blocking cookies entirely may cause some websites to work incorrectly or less effectively.
The use of online tracking mechanisms by third parties is subject to those third parties’ own privacy policies, and not this Policy. If you prefer to prevent third parties from setting and accessing cookies on your computer, you may set your browser to block all cookies. Additionally, you may remove yourself from the targeted advertising of companies within the Network Advertising Initiative by opting out here, or of companies participating in the Digital Advertising Alliance program by opting out here.