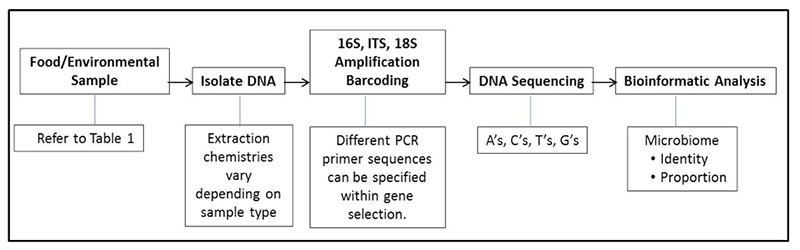
Recall that in article one of this series we wrote that there are two main techniques to obtain a microbiome, a targeted (e.g., bacteria or fungi) or a metagenome (in which all DNA in a sample is sequenced, not just specific targets like bacteria or fungi). In this column we will now explore metagenomes and some applications to food safety and quality.
We have invited Dr. Nur Hasan of CosmosID, Inc., an expert in the field of microbial metagenomics, to share his deep knowledge of metagenomics. Our format will be an interview style.
Safe food production and preservation is a balancing act between food enzymes and microbes. We will start with some general questions about the microbial world, and then proceed deeper into why and how tools such as metagenomics are advancing our ability to explore this universe. Finally, we will ask Dr. Hasan how he sees all of this applying to food microbiology and safe food production.
Greg Siragusa/Doug Marshall: Thank you for joining us. Dr. Hasan, please give us a brief statement of your background and current position.
Nur Hasan: Thanks for having me. I am a molecular biologist by training. I did my bachelor and masters in microbiology, M.B.A in marketing, and Ph.D. in molecular biology. My current position is vice president and head of research and development at CosmosID, Inc., where I am leading the effort on developing the world’s largest curated genome databases and ultra rapid bioinformatics tools to build the most comprehensive, actionable and user-friendly metagenomic analysis platform for both pathogen detection and microbiome characterization.
Siragusa/Marshall: The slogan for CosmosID is “Exploring the Universe of Microbes”. What is your estimate of the numbers of bacterial genera and species that have not yet been cultured in the lab?
Hasan: Estimating the number of uncultured bacteria on earth is an ongoing challenge in biology. The widely accepted notion is more than 99% of bacteria from environmental samples remain ‘unculturable’ in the laboratory; however, with improvements in media design, adjustment of nutrient compositions and optimization of growth conditions based on the ecosystem these bacteria are naturally inhabiting, scientists are now able to grow more bacteria in the lab than we anticipated. Yet, our understanding is very scant on culturable species diversity across diverse ecosystems on earth. With more investigators using metagenomics tools, many ecosystems are being repeatedly sampled, with ever more microbial diversity revealed. Other ecosystems remain ignored, so we only have a skewed understanding of species diversity and what portion of such diversity is actually culturable. A report from Schloss & Handelsman highlighted the limitations of sampling and the fact that it is not possible to estimate the total number of bacterial species on Earth.1 Despite the limitation, they took a stab at the question and predicted minimum bacterial species richness to be 35,498. A more recent report by Hugenholtz estimated that there are currently 61 distinct bacterial phyla, of which 31 have no cultivable representatives.2 Currently NCBI has about 16,757 bacterial species listed, which represent less than 50% of minimum species richness as predicted by Schloss & Handelsman and only a fraction of all global species richness of about 107 to 109 estimated by Curtis and Dykhuizen.3,4
Siragusa/Marshall: In generic terms what exactly is a metagenome? Also, please explain the meaning of the terms “shotgun sequencing”, “shotgun metagenomes”, and “metagenomes”. How are they equivalent, similar or different?
Hasan: Metagenome is actually an umbrella term. It refers to the collection of genetic content of all organisms present in a given sample. It is studied by a method called metagenomics that involves direct sequencing of a heterogeneous population of DNA molecules from a biological sample all at once. Although in most applications, metagenome is often used to refer to microbial metagenome (the genes and genomes of microbial communities of given sample), in a broader sense, it actually represents total genetic makeup of a sample including genomes and gene sequences of other materials in the sample, such as nucleic acids contributed by other food ingredients of plant and animal origin. The metagenome provides an in-depth understanding of the composition, structure, functional and metabolic activities of food, agricultural and human communities.
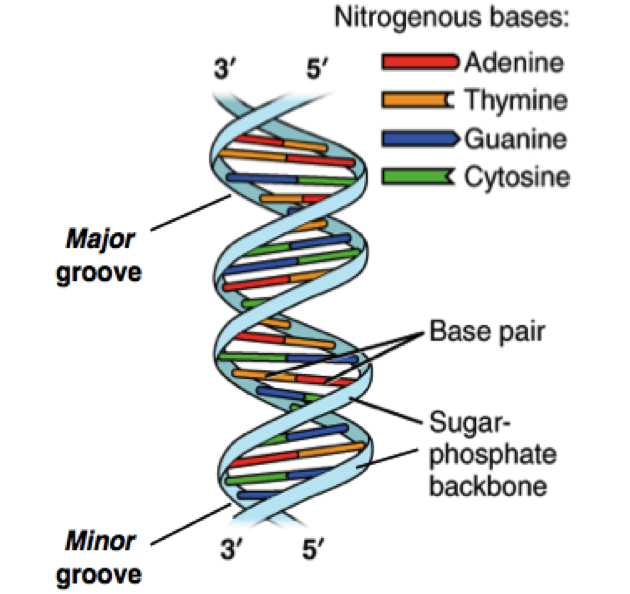
Shotgun sequencing is a method where long strands of DNA (such as an entire genome of a bacterium) are randomly shredded (“shotgunning”) into smaller DNA fragments, so that they can be sequenced individually. Once sequenced, these small fragments are then assembled together into contigs by computer programs that find overlaps in the genetic code, and the complete sequence of the bacterial genome is generated. Now, instead of one genome, if you directly sequence entire assemblage of genomes from a metagenome using such shotgun approach, it’s called shotgun metagenomics and resulting output is termed a shotgun metagenome. By this method, you are literally sequencing thousands of genomes simultaneously from a given metagenome in one assay and get the opportunity to reconstruct individual genomes or genome fragments for investigation and comparison of the genetic consortia and taxonomic composition of complete communities and their predicted functions. Whereas targeted 16S rRNA or targeted 16S amplicon sequencing relies on amplification and sequencing of one target region, the 16S gene region, shotgun metagenomics is actually target free, it is aimed at sequencing entire genomes of every organism present in a sample and gives a more accurate, and unbiased biological representation of a sample. As an analogy of shotgun metagenomics, lets think about your library where you may have multiple books (like as different organisms present in a metagenome). You can imagine shotgun metagenomics as a process whereby all books from your library are shredded, mixed up, and then you will assemble the small shredded pieces to find text overlap and piecing the cover of all books together to reassemble each of your favorite books. Shotgun metagenomics approximates this analogy.
Metagenome and metagenomics are often used interchangeably. Where metagenome is the total collection of all genetic material from a given samples, metagenomics is the method to obtain a metagenome that utilizes a shotgun sequencing approach to sequence all these genetic material at once.
Shotgun sequencing and shotgun metagenomics are also used interchangeably. Shotgun sequencing is a technique where you fragment large DNA strands into small pieces and sequence all small fragments. Now, if you apply such techniques to sequence a metagenome, than we call it shotgun metagenomics.
Go to page 2 of the interview below.